
What is in this leaflet
This leaflet answers some common questions about Rozlytrek. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Rozlytrek against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Rozlytrek is used for
Rozlytrek is used to treat adults with a type of lung cancer called non-small cell lung cancer. It is used if your cancer:
- is 'ROS1-positive' – this means your cancer cells have an alteration in a gene called ROS1 and
- is advanced or has spread to another part of your body (metastatic)
Rozlytrek is also used to treat children (12 years and older), adolescents and adults with cancer that is ‘NTRK fusion-positive’. This means that your cancer cells have an alteration in one of the NTRK genes. Rozlytrek is used when the cancer is advanced or has spread to another part of your body (metastatic) and other treatments have not worked or are not suitable for you.
Rozlytrek contains the active ingredient entrectinib.
Rozlytrek belongs to a group of medicines called anti-neoplastic (or anti-cancer) agents which are used to treat cancer.
Rozlytrek works by blocking the action of enzymes which have an alteration in them. This is due to the alteration in the NTRK or ROS1 genes that make them. The altered enzymes encourage the cancer cells to grow.
Rozlytrek may slow down or stop the cancer growing. It may also help to shrink your cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
Before you take Rozlytrek
When you must not take it
Do not take Rozlytrek if you have an allergy to:
- any medicine containing entrectinib
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed if you are taking this medicine. It is not known if entrectinib passes into breast milk. There is a possibility that your baby may be affected.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- heart problems such as a conditional called ‘prolonged QT interval’. This is shown on an electrocardiogram (ECG). Or a condition called 'congestive heart failure'.
- heart failure – an inability for your heart to adequately pump blood to supply oxygen to the body
- an inherited problem called ‘galactose intolerance’, ‘congenital lactase deficiency’ or ‘glucose-galactose malabsorption’. Rozlytrek contains lactose (a type of sugar). If you have been told by your doctor that you cannot tolerate or digest some sugars, talk to your doctor before taking this medicine.
- liver or kidney problems
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
Rozlytrek may be harmful to an unborn baby when taken by a pregnant woman. You should not take Rozlytrek while you are pregnant.
If you are a woman who could become pregnant, use highly effective contraception (birth control) during treatment, and for at least 5 weeks after taking the last capsule.
If you are the male partner of a woman who could become pregnant, use highly effective contraception during treatment, and for at least 3 months after taking the last capsule.
Talk to your doctor about the right methods of contraception for you and your partner.
Tell your doctor if you are planning to start breast-feeding. It is not recommended that you breastfeed while taking Rozlytrek.
If you have not told your doctor about any of the above, tell him/ her before you start taking Rozlytrek.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Rozlytrek and other medicines may interfere with each other. These include:
- nilotinib, topotecan, lapatinib and mitoxantrone used to treat cancer
- everolimus used to treat cancer or used to prevent the body’s immune system from rejecting a transplanted organ
- sirolimus used to prevent the body’s immune system from rejecting a transplanted organ
- St. John’s Wort, a herbal medicine for depression
- ritonavir and saquinavir used to treat AIDS/HIV
- ketoconazole, itraconazole, voriconazole and posaconazole used to treat fungal infections
- phenytoin, carbamazepine and phenobarbital, anti-epileptics used to treat seizures or fits
- rifampicin and rifabutin used to treat tuberculosis and other infectious diseases
These medicines may be affected by Rozlytrek or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take Rozlytrek
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
How much to take
Your doctor will tell you how many capsules you need to take each day. This may depend on your condition, whether you are taking any other medicines and whether you experience side effects.
For adults, the normal dose of Rozlytrek is three 200 mg capsules taken daily (total dose 600 mg).
For children and adolescents, the dose will be calculated by the doctor based on the patient's weight and height.
How to take it
Swallow the capsules whole with a full glass of water. Do not open or dissolve the capsules.
The capsules can be taken with or without food.
If you vomit immediately after taking a dose of Rozlytrek, take another dose.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
Do not stop taking this medicine without talking to your doctor first.
It is important to take Rozlytrek every day for as long as your doctor prescribes it for you.
If you forget to take it
If it is more than 12 hours until your next dose, take the missed dose as soon as you remember.
If it is less than 12 hours until your next dose, do not take the missed dose. Then take your next dose at the usual time.
Do not take a double dose to make up for the dose that you missed.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (Australia - telephone 13 11 26; New Zealand - telephone 0800 764 766 or 0800 POISON) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Rozlytrek. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Keep telephone numbers for these places handy.
If you are not sure what to do, contact your doctor or pharmacist.
While you are taking Rozlytrek
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Use highly effective contraception to prevent pregnancy while you are being treated with Rozlytrek.
Women must avoid pregnancy during treatment with Rozlytrek and for at least 5 weeks after taking the last capsule. Men must avoid fathering a child during treatment with Rozlytrek and for at least 3 months after taking the last capsule.
If you or your partner become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor will monitor your progress and check for side effects. If necessary, your doctor may decide to reduce your dose, temporarily interrupt your treatment or stop it altogether.
Things you must not do
Do not take Rozlytrek to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Rozlytrek affects you. This medicine may cause confusion, hallucinations, fainting, blurred vision or dizziness in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous. Children should be careful when riding bicycles or climbing trees.
Do not drink grapefruit juice or eat grapefruit during your treatment with Rozlytrek. It may increase the amount of the medicine in your blood to a harmful level.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Rozlytrek.
This medicine helps most people with cancer, but it may have unwanted side effects. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- feeling tired
- pain including headache or head pain, or joint or muscle pain or pain or discomfort in limbs or bones
- fever
- constipation
- diarrhoea
- feeling sick (nausea) or being sick (vomiting), or stomach pain
- difficulty in swallowing
- changes in taste
- an abnormal or unpleasant sense of touch
- numbness or weakness of the arms and legs
- loss of muscle coordination, being unsteady when walking
- symptoms of anaemia such as tiredness, headaches, being short of breath when exercising, dizziness and looking pale
- weight gain
- loss of appetite
- dehydration
- muscle weakness
- bone fractures
- lung infection
- urinary tract infection
- blurred vision
- rash
- swelling or puffiness of the skin
- disturbances in your sleep pattern
If any of the following happen, tell your doctor immediately:
- signs of heart problems (heart failure) such as persistent coughing or wheezing, shortness of breath, and swelling in your legs or arms (fluid retention)
- feeling dizzy or light-headed as this may be a sign of an abnormal heart rhythm or low blood pressure
- feeling confused, changes in mood, having memory problems or seeing things that are not there (hallucinations)
- loss of consciousness or fainting
- symptoms of a condition called tumour lysis syndrome, including nausea or vomiting, muscle cramps or twitches, decreased urination, irritability, sudden uncontrolled fits (seizures).
Your doctor may lower your dose, stop your treatment for a short time or stop your treatment completely.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
Some of these side effects can only be found when your doctor does tests from time to time to check your progress (for example, decreased while blood cell count, increase in blood creatinine, increase in blood liver enzymes, increase uric acid in blood).
After using Rozlytrek
Storage
Keep the capsules in the bottle until it is time to take them. If you take the capsules out of the bottle they may not keep well.
Keep the bottle tightly closed to protect from moisture. Store in a cool dry place where the temperature stays below 30°C.
Do not store Rozlytrek or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Rozlytrek 100 mg capsules are yellow with “ENT 100” imprinted in blue on the side. Rozlytrek 100 mg capsules are supplied in bottles of 30 capsules.
Rozlytrek 200 mg capsules are orange with “ENT 200” imprinted in blue on the side. Rozlytrek 200 mg capsules are supplied in bottles of 90 capsules.
Ingredients
Rozlytrek capsules contain 100 mg or 200 mg of entrectinib as the active ingredient.
The capsules also contain:
- lactose
- microcrystalline cellulose
- tartaric acid
- hypromellose
- crospovidone
- magnesium stearate
- silicon dioxide
The capsule shell contains:
- hypromellose
- titanium dioxide
- iron oxide yellow (100 mg capsule only)
- sunset yellow FCF (200 mg capsule only)
The printing ink contains:
- shellac
- propylene glycol
- strong ammonia solution
- indigo carmine aluminium lake
This medicine does not contain sucrose or gluten.
Manufacturer
Rozlytrek is distributed in Australia by:
Roche Products Pty Limited
ABN 70 000 132 865
Level 8, 30 - 34 Hickson Road
Sydney NSW 2000
AUSTRALIA
Medical enquiries: 1800 233 950
Please check with your pharmacist for the latest Consumer Medicine Information.
Australian Registration Numbers:
100 mg AUST R 318003
200 mg AUST R 318002
Rozlytrek is distributed in New Zealand by:
Roche Products (NZ) Limited
PO Box 109113 Newmarket
Auckland 1149
NEW ZEALAND
Medical enquiries: 0800 276 243
This leaflet was prepared on 11 February 2022
Published by MIMS April 2022

 Recommended dose modifications for specific adverse reactions are provided in Table 3. See Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects).
Recommended dose modifications for specific adverse reactions are provided in Table 3. See Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects).
 Clinically relevant adverse reactions that occurred in ≤ 10% of patients include dysphagia (10%), fall (8%), pleural effusion (8%), fractures (6%), hypoxia (4.2%), pulmonary embolism (3.9%), syncope (3.9%), congestive heart failure (3.4%), and QT prolongation (3.1%).
Clinically relevant adverse reactions that occurred in ≤ 10% of patients include dysphagia (10%), fall (8%), pleural effusion (8%), fractures (6%), hypoxia (4.2%), pulmonary embolism (3.9%), syncope (3.9%), congestive heart failure (3.4%), and QT prolongation (3.1%). Clinically relevant adverse reactions in < 20% of paediatric patients who received Rozlytrek included pruritus, urinary incontinence, eye pain, photophobia, abdominal pain, urinary tract infection and nasal congestion.
Clinically relevant adverse reactions in < 20% of paediatric patients who received Rozlytrek included pruritus, urinary incontinence, eye pain, photophobia, abdominal pain, urinary tract infection and nasal congestion. Table 7 summarises the laboratory abnormalities in STARTRK-NG (n=68), TAPISTRY (n=6) and STARTRK-2 (n=2) in paediatric patients treated with Rozlytrek.
Table 7 summarises the laboratory abnormalities in STARTRK-NG (n=68), TAPISTRY (n=6) and STARTRK-2 (n=2) in paediatric patients treated with Rozlytrek. Other clinically relevant laboratory abnormalities in patients who received Rozlytrek included decreased phosphorous.
Other clinically relevant laboratory abnormalities in patients who received Rozlytrek included decreased phosphorous. Of the 92 patients with ROS1-positive NSCLC in the efficacy evaluable analysis set, 10 had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months prior to study entry. Responses in intracranial lesions were observed in 7 of these 10 patients.
Of the 92 patients with ROS1-positive NSCLC in the efficacy evaluable analysis set, 10 had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months prior to study entry. Responses in intracranial lesions were observed in 7 of these 10 patients.

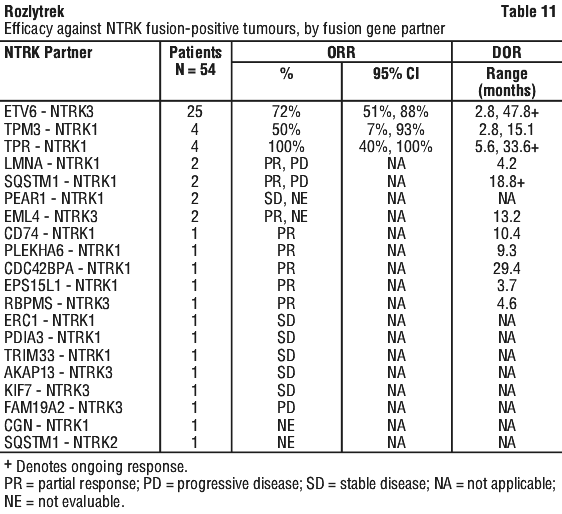 Among the subset of patients who received prior systemic therapy for metastatic disease, the ORR was 53%, similar to that seen in the overall population.
Among the subset of patients who received prior systemic therapy for metastatic disease, the ORR was 53%, similar to that seen in the overall population.