1 Name of Medicine
Fostemsavir trometamol.
2 Qualitative and Quantitative Composition
Each film-coated tablet contains 600 mg of fostemsavir (as fostemsavir trometamol).
List of excipients with known effect.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Extended release tablets.
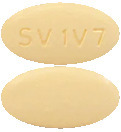
Beige, film-coated, biconvex, oval tablets which may have a slight odour (vinegar-like), debossed with 'SV 1V7' on one side.
4.1 Therapeutic Indications
Rukobia is indicated in combination with other antiretroviral agents for the treatment of heavily treatment-experienced adults with multidrug-resistant human immunodeficiency virus-1 (HIV-1) infection for whom it is otherwise not possible to construct a suppressive anti-viral regimen due to resistance, intolerance or safety considerations (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
4.2 Dose and Method of Administration
Therapy should be initiated by a physician experienced in the management of HIV infection.
Fostemsavir can be taken with or without food.
Fostemsavir tablets should be swallowed whole, and should not be chewed, crushed or split.
Adults.
The recommended dosage of fostemsavir is 600 mg orally twice daily.
Adolescents and children.
Fostemsavir is not recommended in children below 18 years of age due to a lack of safety and efficacy data.
Elderly.
There are limited data available on the use of fostemsavir in patients aged 65 years and older. However, there is no evidence that elderly patients require a different dose than younger adult patients (see Section 5.2 Pharmacokinetic Properties, Special patient populations).
Renal impairment.
No dosage adjustment of fostemsavir is required for patients with renal impairment and those on haemodialysis (see Section 5.2 Pharmacokinetic Properties, Special patient populations).
Hepatic impairment.
No dosage adjustment is required in patients with hepatic impairment (see Section 5.2 Pharmacokinetic Properties, Special patient populations).4.3 Contraindications
Fostemsavir is contraindicated in patients who have demonstrated hypersensitivity to fostemsavir or any components of formulations of fostemsavir.
Fostemsavir is contraindicated in combination with strong CYP3A inducers including, but not limited to: carbamazepine, phenytoin (anticonvulsants), mitotane (antineoplastic), enzalutamide (androgen receptor inhibitor), rifampicin (antimycobacterial) and St John's wort (Hypericum perforatum, herbal supplement) (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
4.4 Special Warnings and Precautions for Use
Immune reconstitution syndrome.
In HIV-infected patients with severe immune deficiency at the time of initiation of antiretroviral therapy (ART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of ART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections and Pneumocystis jiroveci (P. carinii) pneumonia. Any inflammatory symptoms must be evaluated without delay and treatment initiated when necessary. Autoimmune disorders (such as Graves' disease, polymyositis and Guillain-Barre syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment and sometimes can be an atypical presentation.
QTc prolongation.
In healthy study participants, a supratherapeutic dose of fostemsavir (2400 mg twice daily) has been shown to significantly prolong the QTc interval of the electrocardiogram (see Section 5.1 Pharmacodynamic Properties, Pharmacodynamic effects). Fostemsavir should be used with caution in patients with a history of QT interval prolongation, when co-administered with a drug with a known risk of Torsade de Pointes (e.g. amiodarone, disopyramide, dofetilide, ibutilide, procainamide, quinidine, or sotalol) or in patients with relevant pre-existing cardiac disease. Elderly patients may be more susceptible to drug-induced QT interval prolongation.
Patients with hepatitis B or C virus co-infection.
Monitoring of liver chemistries is recommended in patients with hepatitis B and/or C coinfection. Particular diligence should be applied in initiating or maintaining effective hepatitis B therapy (referring to treatment guidelines) when starting fostemsavir therapy in HIV-hepatitis B co-infected patients.
Opportunistic infections.
Patients receiving fostemsavir or any other antiretroviral therapy may still develop opportunistic infections and other complications of HIV infection. Therefore, patients should remain under close clinical observation by physicians experienced in the treatment of these associated HIV diseases.
Transmission of infection.
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Drug interactions.
Caution should be given to co-administering medications (prescription and non-prescription) that may change the exposure to temsavir, the active moiety of fostemsavir, or medications that may have their exposure changed by temsavir (see Section 4.3 Contraindications; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Reduced exposure to temsavir may lead to loss of therapeutic effect of Rukobia and possible development of resistance (see Section 4.3 Contraindications). Increased exposure to temsavir may increase the risk of QTc interval prolongation (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties, Pharmacodynamic effects).
Co-administration of fostemsavir with elbasvir/grazoprevir is not recommended as increased grazoprevir concentrations may increase the risk of ALT elevations (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Dose modifications and/or careful titration of dose is recommended for certain statins that are substrates of OATP1B1/3 or BCRP (rosuvastatin, atorvastatin, pitavastatin, simvastatin and fluvastatin) when co-administered with fostemsavir (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
When fostemsavir was co-administered with oral contraceptives, temsavir increased concentrations of ethinyl estradiol and caution is advised particularly in patients with additional risk factors for thromboembolic events. Doses of estrogen-based therapies, including oral contraceptives, should not contain more than 30 microgram of ethinyl estradiol per day in patients who are receiving fostemsavir (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Use in the elderly.
See Section 4.2 Dose and Method of Administration, Elderly.
Paediatric use.
Fostemsavir is not recommended in children below 18 years of age due to a lack of safety and efficacy data.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of fostemsavir on the pharmacokinetics of other agents.
Significant interactions are not expected when fostemsavir is co-administered with substrates of cytochrome P450 (CYPs), uridine diphosphate glucuronosyl transferases (UGTs), P-glycoprotein (P-gp), multidrug resistance protein (MRP)2, bile salt export pump (BSEP), sodium taurocholate co-transporting polypeptide (NTCP), organic anion transporters (OAT)1, OAT3, organic cation transporters (OCT)1, and OCT2 based on in vitro and clinical drug interaction data.
In vitro, temsavir was not a clinically relevant inhibitor of other transporters, major CYP enzymes or UGTs (IC50 > 40 microM). In addition, temsavir did not induce CYP enzymes in vitro.
In vitro, temsavir inhibited organic anion transporter polypeptides (OATP)1B1 and OATP1B3 (IC50 = 32 and 16 microM, respectively). Additionally, temsavir and its two metabolites (BMS-646915 and BMS-930644) inhibited breast cancer resistance protein (BCRP) (IC50 = 12, 35, and 3.5 to 6.3 microM, respectively). Based on these data, temsavir is expected to affect the pharmacokinetics of drugs that are substrates of OATP1B1/3 or BCRP (e.g. rosuvastatin, atorvastatin, simvastatin, pitavastatin and fluvastatin). Therefore, dose modifications and/or careful titration of dose is recommended for certain statins.
Based on in vitro data, temsavir and its two metabolites (BMS-930644 and BMS-646915) inhibited multidrug and toxin extrusion protein (MATE)1/2K. However, this interaction is unlikely to be of clinical significance.
BMS-930644, a metabolite of temsavir, inhibited CYP3A4, BCRP, MATE2K, and OCT1 with IC50 values < 10 microM. However, as circulating concentrations of BMS-930644 are low [Cmax of approximately 458 nanogram/mL (~1 microM) with fostemsavir 600 mg twice daily], clinically significant interactions are unlikely.
Effect of other agents on the pharmacokinetics of temsavir.
Temsavir is a substrate of CYP3A, esterases, P-gp and BCRP, but not of OATP1B1 or OATP1B3. Its biotransformation to two circulating metabolites, BMS-646915 and BMS-930644, is mediated by unidentified esterases (36.1%) and by CYP3A4 enzyme (21.2%), respectively. Temsavir exposures may be influenced by modulators of CYP3A4, P-gp and/or BCRP activity. However, because of the primary esterase metabolism pathway, effects are expected to be less than that of substrates primarily metabolized by CYP3A4. When fostemsavir was co-administered with a strong CYP3A inducer rifampicin, a significant reduction in temsavir plasma concentrations was observed. Significant decreases in temsavir plasma concentrations may also occur when fostemsavir is co-administered with other strong CYP3A inducers and may result in loss of virologic response (see Section 4.3 Contraindications).
Fostemsavir may be co-administered with strong CYP3A4, BCRP and/or P-gp inhibitors (e.g. clarithromycin, itraconazole, posaconazole, and voriconazole) without dose adjustment based on the results of clinical drug interaction studies with cobicistat and ritonavir.
Selected drug interactions are presented in Table 1. Recommendations are based on either drug interaction studies or predicted interactions due to the expected magnitude of the interaction and/or potential for serious adverse events or loss of efficacy.


4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the effects of fostemsavir on human male or female fertility. Animal studies indicate no effects of fostemsavir on male or female fertility at clinically relevant doses. Oral administration of fostemsavir had no adverse effects on male or female fertility in rats at doses up to 300 mg/kg/day in males and 600 mg/kg/day in females (≥ 33 times the 600 mg twice daily human clinical exposure based on AUC). Effects in males included dose-dependent gross and microscopic pathological findings in the testes and epididymides, reductions in prostate gland/seminal vesicle weights, and decreased sperm density (at ≥ 21 times the 600 mg twice daily human clinical exposure based on AUC), with decreased motility and increased abnormal sperm (at ≥ 25 times the 600 mg twice daily human clinical exposure based on AUC). These findings were not considered clinically relevant.
(Category B3)
There are insufficient data to support fostemsavir use in pregnancy. The Antiviral Pregnancy Registry (APR) monitors for birth defects in neonates and other pregnancy outcomes in individuals exposed to fostemsavir during pregnancy in an observational setting. The effect of fostemsavir on human pregnancy is unknown.
Animal studies indicate no effects of fostemsavir on embryo-fetal development at clinically relevant exposures. Fostemsavir was associated with developmental toxicity findings in animal reproductive studies at exposures substantially higher than the therapeutic dose in the presence of maternal toxicity.
No fetal abnormalities were observed following oral administration of fostemsavir to pregnant rats during organogenesis at 600 mg/kg/day (52 times the predicted human exposure at the maximum recommended human dose (MRHD)). No adverse effects were observed on pregnancy, delivery or fetal and early offspring development when fostemsavir was administered at oral doses up to 300 mg/kg/day through pregnancy and lactation (37 times the human exposure at the MRHD). However, oral administration of fostemsavir to pregnant rats did result in fetal abnormalities (cleft palate, open eyes, shortened snout, microstomia, misaligned mouth/jaw and protruding tongue) and reductions in fetal body weights in the presence of maternal toxicity (reductions in body weights and food consumption) when dosed at 1000 mg/kg/day (65 times the predicted human exposure at the MRHD).
No adverse effects on embryonic survival and fetal weights were evident following oral administration of fostemsavir to pregnant rabbits during organogenesis at 50 mg/kg/day (≥ 3 times the predicted human exposure at the MRHD). Decreases in fetal body weights and embryonic deaths were evident at 6 times the exposure at the MRHD. At doses equal to or greater than 250 mg/kg/day (13 times the exposures at MRHD), oral administration of fostemsavir to pregnant rabbits resulted in abortions in the presence of severe maternal toxicity (deaths and inappetence, body weight loss).
Temsavir was shown to cross the placenta in an animal distribution study after dosing with radio-labelled fostemsavir.
Fostemsavir should be used during pregnancy only if the expected benefit justifies the potential risk to the fetus.
Health experts recommend that where possible, HIV-infected women do not breast feed their infants in order to avoid transmission of HIV. In settings where formula feeding is not feasible, local official lactation and treatment guidelines should be followed when considering breast feeding during antiretroviral therapy.
It is expected that temsavir will be secreted into human milk based on animal data, although this has not been confirmed in humans. In a distribution study, fostemsavir-related drug materials (i.e. temsavir and/or temsavir-derived metabolites) were excreted in rat milk following a single dose of fostemsavir administered to lactating rats 7 to 9 days postpartum. In the pre-and postnatal development study in rats, temsavir was present in milk at concentrations similar to those measured in maternal plasma, as determined 11 days postpartum.
In a pre- and postnatal development study in rats, lactational exposure at 300 mg/kg/day (corresponding to a plasma exposure multiple 35 times that in humans at 600 mg twice daily based on AUC) was associated with reduced neonatal survival from post-natal days 7 to 14. Therefore, it is recommended that when possible, HIV-infected women should not breast feed while receiving fostemsavir.4.7 Effects on Ability to Drive and Use Machines
There have been no studies to investigate the effect of fostemsavir on driving performance or the ability to operate machinery. The clinical status of the patient and the adverse event profile of fostemsavir should be borne in mind when considering the patient's ability to drive or operate machinery.
4.8 Adverse Effects (Undesirable Effects)
Clinical trial data.
A total of 620 HIV-1 infected subjects received at least one dose of fostemsavir as part of a controlled clinical trial.
The safety and tolerability of the recommended dose of fostemsavir was evaluated in a Phase III, partially-randomised, double-blind, placebo-controlled trial (BRIGHTE [205888]) conducted in 371 heavily treatment-experienced adult subjects (see Section 5.1 Pharmacodynamic Properties, Clinical trials). In the Randomised Cohort, 272 subjects received either blinded fostemsavir, 600 mg twice daily (n = 203), or placebo (n = 69), in addition to their current failing regimen, for 8 days of functional monotherapy. Beyond Day 8, randomised subjects received open-label fostemsavir, 600 mg twice daily, plus an optimised background therapy (OBT). In the Non-randomised Cohort, 99 subjects received open-label fostemsavir, 600 mg twice daily, plus OBT from Day 1 onward.
Adverse drug reactions (ADRs) identified in the Phase III clinical trial, which included a total of 370 subjects receiving at least 1 dose of fostemsavir 600 mg twice daily, are listed in Table 2 by MedDRA system organ class and by frequency. Frequencies are defined as: very common (≥ 1/10), common (≥ 1/100 and < 1/10), uncommon (≥ 1/1,000 and < 1/100), rare (≥ 1/10,000 and < 1/1,000) and very rare (< 1/10,000), including isolated reports. For many of the adverse drug reactions listed, it is unclear whether they are related to fostemsavir, or the other medicinal products used in the management of HIV infection, or whether they are a result of the underlying disease process.

Changes in laboratory chemistries.
Increases in creatine phosphokinase (CPK) were observed following treatment with fostemsavir, which were mainly mild or moderate. These changes were rarely associated with musculoskeletal complaints and are not considered clinically relevant.
Clinically relevant increases in serum creatinine have primarily occurred in patients with identifiable risk factors for reduced renal function, including pre-existing medical history of renal disease and/or concomitant medications known to cause increases in creatinine. A causal association between fostemsavir and elevation in serum creatinine has not been established.
Increases in direct (conjugated) bilirubin have been observed following treatment with fostemsavir. Cases of clinical significance were uncommon and were confounded by the presence of intercurrent serious comorbid events not related to dosing with study medication (e.g. sepsis, cholangiocarcinoma or other complications of viral hepatitis co-infection). In the remaining reports, elevations in direct bilirubin (without clinical jaundice) were typically transient, occurred without increases in liver transaminases and resolved on continued fostemsavir. In vitro, temsavir and its metabolites inhibit OATP1B1 and OATP1B3; two well-recognized transporters of direct and indirect (unconjugated) bilirubin (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Fostemsavir may contribute to elevations in bilirubin when co-administered with other drugs known to cause hyperbilirubinemia, or when dosed in patients with liver disease or who otherwise have reduced activity of hepatic transport proteins, including patients with HIV infection.
Post-marketing data.
No data available.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Symptoms and signs.
There is currently limited experience of overdosage with fostemsavir.
Treatment.
There is no specific treatment for overdose with fostemsavir. If overdose occurs, the patient should be treated supportively with appropriate monitoring as necessary. As temsavir is highly bound to plasma proteins, it is unlikely that it will be significantly removed by dialysis.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Fostemsavir is a prodrug without significant biochemical or antiviral activity that is hydrolyzed to the active moiety, temsavir, upon cleavage of a phosphonooxymethyl group in vivo. Temsavir binds directly to the gp120 subunit within the HIV-1 envelope glycoprotein gp160 and selectively inhibits the interaction between the virus and cellular CD4 receptors, thereby preventing viral entry into, and infection of, host cells. Temsavir inhibited the binding of soluble CD4 to surface immobilized gp120 with an IC50 of 14 to 30 nanoM using an enzyme-linked immunosorbent assay (ELISA).
Pharmacodynamic effects.
Antiviral activity in cell culture.
Temsavir exhibited antiviral activity against CCR5-tropic (n = 3; EC50 range 0.4 to 1.7 nanoM), CXCR4-tropic (n = 5; EC50 range 0.7 to > 2,000 nanoM) and dual/mixed-tropic (n = 1; EC50 58 nanoM) laboratory strains of subtype B HIV-1.
In one study, a total of 103 clinical isolates were examined for susceptibility to temsavir using PBMCs as the host cell. These viruses spanned multiple Group M subtypes, including A, B, B', C, D, F, CRF01_AE and G. In addition, 2 Group O viruses and 1 HIV-2 virus were tested for temsavir susceptibility. The cohort contained mostly CCR5-tropic viruses, but there were also some CXCR4-tropic and dual-tropic strains. For most of the subtypes, temsavir exhibited potent but variable activity regardless of tropism. However, all nine viruses examined from subtype CRF01_AE, both viruses examined from Group O and one HIV-2 virus all displayed reduced susceptibility to temsavir at the highest concentration tested.
In another study, a total of 1337 isolates have been examined to date in the PhenoSense Entry Assay. These include viruses from all subjects in the Phase IIa (206267), Phase IIb (205889) and Phase III (205888) studies, plus other samples obtained from plasma samples of infected individuals. A total of 881 of these samples were from subtype B, 156 samples from subtype C, 43 samples from subtype A, 17 samples from subtype A1, 48 samples from subtype F1, 29 samples from subtype BF1 and 19 samples from subtype BF infected individuals. In addition, there were 5 CRF01_AE samples: four of these samples exhibited IC50 values above the maximum concentration of the assays used (100 nanoM or 10 microM), while one sample exhibited an IC50 of ~222 nanoM. Of all isolates tested, 53.8% and 80.1% exhibited IC50s < 1 nanoM, and < 10 nanoM, respectively, for all subtypes. Each of the subtypes displayed wide ranges of susceptibility to temsavir. For the subtype B viruses, IC50s ranged from the low picoM to > 10 microM. The other subtypes had similar ranges. Geometric mean IC50s ranged from 1.15 nanoM for subtype B virus to 34.91 nanoM for the BF1 subtype. These results demonstrate there is a large range of intrinsic susceptibility to temsavir in pre-treatment envelopes within the population.
Antiviral activity in combination with other antiviral agents.
No drugs with inherent anti-HIV activity were antagonistic with temsavir (in vitro assessments were performed in combination with abacavir, didanosine, zalcitabine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil fumarate, zidovudine, efavirenz, nevirapine, amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, enfuvirtide, maraviroc, ibalizumab, delavirdine, rilpivirine, darunavir, dolutegravir or raltegravir). In addition, antivirals without inherent anti-HIV activity (entecavir, ribavirin) have no apparent effect on temsavir activity.
Effect of human serum and serum proteins.
In vitro studies showed no significant serum effect. Infection of PM1 or MT-2 cells with laboratory strains HIV-1 LAI and HIV-1 NL4-3 demonstrated that the presence of 40% human serum decreased the anti-HIV potency of temsavir by 1.5-2.1-fold.
Resistance in vitro.
HIV-1 variants with reduced susceptibility to temsavir were selected in cell culture following passage of NL4-3, LAI and BaL viruses in a T-cell line. Emerging amino acids in gp120 that reduced susceptibility were identified and included L116P/Q, A204D, M426L, M434I, and M475I (S375I/N substitutions were identified based on in vivo data with a related attachment inhibitor).
Single-substitution recombinant viruses were engineered into the HIV-1 LAI viral background, and the resultant recombinants were examined against temsavir. Substitution M426L was associated with an 81-fold decrease in temsavir sensitivity for the recombinant virus, while a M434I or M475I change displayed a moderate effect on sensitivity to the inhibitor (11- and 4.8-fold decrease, respectively). Two other amino acid substitutions, L116P and A204D, located distal to the CD4 binding pocket of gp120, conferred high levels of resistance to temsavir in an LAI background (> 340-fold decrease). However, both amino acids are strictly conserved within clinical envelope genes and these specific polymorphisms at these positions have not been observed clinically during treatment with fostemsavir.
Temsavir remained active against laboratory derived CD4-independent viruses. Treatment with fostemsavir is therefore unlikely to promote resistance to temsavir via generation of CD4 independent virus.
There was no evidence of cross-resistance to representative agents from other ARV classes, including NRTIs, NNRTIs, PIs and INSTIs. Temsavir retained wild-type activity against viruses resistant to tenofovir, abacavir, zidovudine, lamivudine, rilpivirine, efavirenz, atazanavir, darunavir or raltegravir. Additionally, NRTIs (abacavir, tenofovir), NNRTIs (rilpivirine, efavirenz), PIs (atazanavir, darunavir) and the INSTI raltegravir, retained activity against site-directed mutants with reduced temsavir susceptibility (S375M, M426L, or M426L plus M475I).
No cross-resistance was observed between temsavir and maraviroc or enfuvirtide. Temsavir was active against viruses with resistance to enfuvirtide. Some CCR5-tropic, maraviroc-resistant, viruses showed reduced susceptibility to temsavir, however, there was no absolute correlation between maraviroc resistance and reduced sensitivity to temsavir. Maraviroc and enfuvirtide retained activity against clinical envelopes that had reduced susceptibility to temsavir and contained S375H, M426L, or M426L plus M475I substitutions.
Temsavir was active against several ibalizumab-resistant viruses. Ibalizumab retained activity against site-directed mutants that had reduced susceptibility to temsavir (S375M, M426L, or M426L plus M475I). HIV-1 gp120 E202 was identified as a rare treatment-emergent substitution in BRIGHTE that can reduce susceptibility to temsavir, and, depending on the sequence context of the envelope, may also result in reduced susceptibility to ibalizumab.
Resistance in vivo.
Results of the Phase III study (BRIGHTE [205888]) in heavily treatment-experienced adult subjects demonstrated that, overall, virologic response at Day 8 and subsequent timepoints (Weeks 24, 48, and 96) in the Randomised Cohort was not reliably predicted by baseline temsavir IC50-fold change value or the presence of a gp160 substitution of interest.
Temsavir IC50 FC > 100-fold was associated with a median change in HIV-1 RNA from Day 1 to Day 8 of < 0.5 log10 c/mL. Similarly, the presence at baseline of pre-defined gp160 substitutions, identified as potentially important for determining phenotypic susceptibility to temsavir (S375H/I/M/N/T, M426L/P, M434I/K and M475I), was associated with a lower decline in HIV-1 RNA. However, increased baseline temsavir IC50 FC, or the presence of pre-defined gp160 substitutions, did not preclude subjects from achieving a response of > 1 log10 c/mL at Day 8. Indeed, 8 of 21 (38%) subjects with IC50 FC > 100-fold did achieve a Day 8 response > 0.5 log10 c/mL and 7/21 (33%) subjects achieved a > 1 log10 c/mL decline in viral load. Subjects with no pre-defined gp160 substitutions present at baseline achieved a median change in HIV-1 RNA of -1.032 log10 c/mL at Day 8, compared to -0.652 log10 c/mL change in viral load in subjects with pre-defined gp160 substitutions present. Baseline gp160 substitutions most associated with response < 0.5 log10 c/mL at Day 8 were S375H/M/N and M426L.
With the addition of an optimised background therapy, increased baseline temsavir IC50 FC, or the presence of pre-defined gp160 substitutions, did not influence durability of response (HIV-1 RNA < 40 c/mL) through Week 96. These results demonstrate that response to fostemsavir, as determined by baseline virologic factors, is highly context dependent.
The percentage of Randomised subjects who experienced protocol defined virologic failure (PDVF) was 11% (31/272) through Week 24, 18% (49/272) through Week 48, and 23% (63/272) through Week 96. The criteria for PDVF were as follows: HIV-1 RNA confirmed, or last available prior to discontinuation, ≥ 400 c/mL at any time after prior confirmed suppression to < 400 c/mL, or confirmed, or last available, > 1 log10 c/mL increase in HIV-1 RNA at any time above nadir level where nadir is ≥ 40 c/mL (prior to Week 24); HIV-1 RNA confirmed, or last available, ≥ 400 c/mL (at or after Week 24). In the PDVF population through Week 96, 48% (24/50) of evaluable Randomised subjects had treatment-emergent gp160 genotypic substitutions of interest. Most frequently, there was emergence of M426L (32%), S375N (24%), M475I (12%) and M434I (10%). Median increase in temsavir IC50 FC among Randomised subjects meeting PDVF criteria was 1.67-fold; 37% (19/51) of evaluable subjects had temsavir IC50 FC ≤ 10-fold and 49% (25/51) had temsavir IC50 FC ≤ 100-fold at the time of virologic failure, indicating that a proportion of subjects likely retained phenotypic susceptibility to temsavir, despite meeting virologic failure criteria (temsavir IC50 FC is normalised to a 1 nanoM reference virus and is therefore approximately equal to temsavir IC50 expressed in nanoM). Approximately 30% (17/63) of Randomised subjects who met PDVF were subsequently able to achieve virologic suppression to < 40 c/mL.
Reduced antiviral activity against subtype AE.
There were only two subjects with subtype AE virus at screening in the Randomised Cohort. One subject (IC50 FC > 4747-fold and gp160 substitutions at S375H and M475I at baseline) did not respond to fostemsavir at Day 8. A second subject (IC50 FC 298-fold and gp160 substitution at S375N at baseline) received placebo during functional monotherapy. Both subjects were virologically suppressed at Week 96, while receiving fostemsavir plus optimised background therapy.
Effects on electrocardiogram.
In a randomised, placebo- and active-controlled, double-blind, cross-over thorough QT study, 60 healthy subjects received oral administration of placebo, fostemsavir 1200 mg once daily, fostemsavir 2400 mg twice daily and moxifloxacin 400 mg (active control) in random sequence. Fostemsavir administered at 1200 mg once daily did not have a clinically meaningful effect on the QTc interval as the maximum mean time-matched (2-sided 90% upper confidence bound) placebo-adjusted QTc change from baseline based on Fridericia's correction method (QTcF) was 4.3 (6.3) milliseconds (below the clinically important threshold of 10 milliseconds). However, fostemsavir administered at 2400 mg twice daily for 7 days was associated with a clinically meaningful prolongation of the QTc interval as the maximum mean time-matched (2-sided 90% upper confidence bound) for the placebo-adjusted change from baseline in QTcF interval was 11.2 (13.3) milliseconds. Steady-state administration of fostemsavir 600 mg twice daily resulted in a mean temsavir Cmax approximately 4.2-fold lower than the temsavir concentration predicted to increase QTcF interval 10 milliseconds (see Section 4.4 Special Warnings and Precautions for Use).
Clinical trials.
The efficacy of fostemsavir in HIV-infected, heavily treatment-experienced adult subjects is based on data from a Phase III, partially-randomised, international, double-blind, placebo-controlled trial (BRIGHTE [205888]).
The BRIGHTE study was conducted in 371 heavily-treatment experienced HIV-1 infected subjects with multi-class resistance. All subjects were required to have a viral load greater than or equal to 400 copies/mL and ≤ 2 antiretroviral (ARV) classes remaining at baseline due to resistance, intolerability, contraindication, or other safety concerns. At Screening, subjects from the Randomised Cohort had one, but no more than two, fully active and available antiretroviral agents which could be combined as part of an efficacious background regimen. Within the Randomised Cohort, 272 subjects received either blinded fostemsavir, 600 mg twice daily (n = 203), or placebo (n = 69), in addition to their current failing regimen, for 8 days of functional monotherapy. Beyond Day 8, Randomised subjects received open-label fostemsavir, 600 mg twice daily, plus an optimised background therapy (OBT) selected by the Principal Investigator. The Randomised Cohort provides primary evidence of efficacy of fostemsavir. Within the Non-randomised Cohort, 99 subjects with no fully active and approved antiretroviral agents available at Screening, were treated with open-label fostemsavir, 600 mg twice daily, plus OBT from Day 1 onward. The use of an investigational drug(s) as a component of the OBT was permitted in the Non-randomised Cohort.
Overall, the majority of subjects were male (78%), white (70%), and the median age was 49.0 years (range: 17-73 years). At baseline, the median HIV-1 RNA was 4.6 log10 copies/mL and the median CD4+ cell count was 80.0 cells/mm3 (100 and 41 cells/mm3 for Randomised and Non-randomised subjects, respectively). Seventy-five percent (75%) of all treated subjects had a CD4+ T-cell count < 200 cells/mm3 at baseline (with 30% < 20 cells/mm3). Overall, 86% had a history of Acquired Immune Deficiency Syndrome (AIDS), and 8% had a history of hepatitis B and/or C virus co-infection. Seventy one percent (71%) of subjects had been treated for HIV for > 15 years, 85% had been exposed to ≥ 5 different HIV treatment regimens upon entry into the study.
Fifty two percent of subjects in the Randomised Cohort had one fully active agent within their initial OBT, 42% had two, and 6% had zero. Within the Non-randomised Cohort, 81% of subjects had no fully active agents in their original OBT and 19% had one, including 15% (n = 15) who received ibalizumab, which was an investigational agent at the time of BRIGHTE study start-up.
The primary endpoint analysis, based on the adjusted mean decline in HIV-1 RNA from Day 1 at Day 8 in the Randomised Cohort, demonstrated superiority of fostemsavir to placebo (0.79 vs. 0.17 log10 decline, respectively; p < 0.0001, Intent To Treat-Exposed [ITT-E] population) (Table 3).
 At Day 8, 65% (131/203) and 46% (93/203) of subjects had a reduction in viral load from baseline > 0.5 log10 c/mL and > 1 log10 c/mL, respectively, in the fostemsavir group, compared with 19% (13/69) and 10% (7/69) of subjects, respectively, in the placebo group.
At Day 8, 65% (131/203) and 46% (93/203) of subjects had a reduction in viral load from baseline > 0.5 log10 c/mL and > 1 log10 c/mL, respectively, in the fostemsavir group, compared with 19% (13/69) and 10% (7/69) of subjects, respectively, in the placebo group.
By subgroup analysis, fostemsavir-treated Randomised subjects with baseline HIV-1 RNA > 1,000 c/mL achieved a median decline in viral load of 1.02 log10 c/mL at Day 8, compared with 0.00 log10 c/mL decline in subjects treated with blinded placebo.
Virologic outcomes by ITT-E Snapshot Analysis at Weeks 24, 48 and 96 in the BRIGHTE trial (including outcomes by key baseline covariates) are shown in Table 4 for the Randomised Cohort.
 In the Randomised Cohort, viral load < 200 HIV-1 RNA copies/mL was achieved in 68%, 69% and 64% of subjects at Weeks 24, 48 and 96, respectively. At these timepoints, the proportion of subjects with viral load < 400 HIV-1 RNA copies/mL was 75%, 70% and 64%, respectively (ITT-E, Snapshot algorithm). Mean changes in CD4+ T-cell count from baseline continued to increase over time (i.e. 90 cells/mm3 at Week 24, 139 cells/mm3 at Week 48 and 205 cells/mm3 at Week 96). Based on a sub-analysis in the Randomised Cohort, subjects with the lowest baseline CD4+ T-cell counts (< 20 cells/mm3) had a similar increase in CD4+ count over time compared with subjects with higher baseline CD4+ T-cell count (> 50, > 100, > 200 cells/mm3).
In the Randomised Cohort, viral load < 200 HIV-1 RNA copies/mL was achieved in 68%, 69% and 64% of subjects at Weeks 24, 48 and 96, respectively. At these timepoints, the proportion of subjects with viral load < 400 HIV-1 RNA copies/mL was 75%, 70% and 64%, respectively (ITT-E, Snapshot algorithm). Mean changes in CD4+ T-cell count from baseline continued to increase over time (i.e. 90 cells/mm3 at Week 24, 139 cells/mm3 at Week 48 and 205 cells/mm3 at Week 96). Based on a sub-analysis in the Randomised Cohort, subjects with the lowest baseline CD4+ T-cell counts (< 20 cells/mm3) had a similar increase in CD4+ count over time compared with subjects with higher baseline CD4+ T-cell count (> 50, > 100, > 200 cells/mm3).
In the Non-randomised Cohort (subjects with no fully active and approved antiretroviral agents available at screening), HIV-1 RNA < 40 copies/mL was achieved in 37%, 38% and 37% of subjects at Weeks 24, 48 and 96, respectively. At these timepoints, the proportion of subjects with HIV-1 RNA < 200 copies/mL was 42%, 43% and 39%, and the proportion of subjects with HIV-1 RNA < 400 copies/mL was 44%, 44% and 40%, respectively (ITT-E, Snapshot algorithm). Mean changes in CD4+ cell count from baseline increased over time: 41 cells/mm3 at Week 24, 64 cells/mm3 at Week 48 and 119 cells/mm3 at Week 96.
5.2 Pharmacokinetic Properties
The pharmacokinetics of temsavir following administration of fostemsavir are similar between healthy and HIV-infected subjects. Between-subject variability (%CV) in plasma temsavir Cmax and AUC ranged from 22 to 50% and Cτ from 50 to 127% across Phase I studies in healthy subjects. The magnitude of variability was similar in HIV infected subjects (%CV in plasma temsavir Cmax and AUC ranged from 20.5 to 63% and Cτ from 20 to 165%). Between-subject variability in oral clearance and central oral volume of distribution estimated from population pharmacokinetic analysis of healthy subjects from selected Phase I studies and HIV-1 infected patients were 43% and 48%, respectively.
Absorption.
Fostemsavir is a highly soluble prodrug that is metabolized to temsavir by alkaline phosphatase at the luminal surface of the small intestine and is generally not detectable in plasma following oral administration. The active moiety, temsavir, is readily absorbed with the median time to maximal plasma concentrations (Tmax) at 2 hours post dose (fasted). Following oral administration, increases in plasma temsavir exposure (Cmax and AUC) appeared dose proportional, or slightly greater than dose proportional, over the range of 600 mg to 1,800 mg of fostemsavir. Temsavir is absorbed across the small intestine and cecum/proximal ascending colon.
Pharmacokinetic parameters following multiple oral doses of fostemsavir 600 mg twice daily in healthy and HIV-1 infected, heavily-treatment experienced adult subjects are shown in Table 5.
 The absolute bioavailability of temsavir was 26.9% following oral administration of a single 600 mg dose of fostemsavir.
The absolute bioavailability of temsavir was 26.9% following oral administration of a single 600 mg dose of fostemsavir.
Effect of food.
Fostemsavir may be administered with or without food. Temsavir bioavailability (AUC) was not impacted by a standard meal (approximately 423 kcal, 36% fat) but increased 81% with a high-fat meal (approximately 985 kcal, 60% fat) and is not considered clinically significant. Regardless of calorie and fat content, food had no impact on plasma temsavir Cmax. In the Phase 3 BRIGHTE study that demonstrated efficacy and safety, fostemsavir was taken with or without food (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Distribution.
Temsavir is approximately 88% bound to human plasma proteins based on in vivo data. Human serum albumin is the major contributor to plasma protein binding of temsavir in humans. The volume of distribution of temsavir at steady state (Vss) following intravenous administration is estimated at 29.5 L. The blood-to-plasma total radiocarbon Cmax ratio was approximately 0.74, indicating minimal association of temsavir or its metabolites with red blood cells. Ex vivo, the blood-to-plasma ratio (determined at 300, 1000, and 10,000 nanogram/mL) ranged from 0.785 to 0.963 [overall mean (SD) 0.869 ± 0.0599] with no apparent concentration dependence in the concentration range tested. Free fraction of temsavir in plasma was approximately 12 to 18% in healthy subjects, 23% in subjects with severe hepatic impairment, and 19% in subjects with severe renal impairment, and 12% in HIV-1 infected patients.
Metabolism.
In vivo, temsavir is primarily metabolised via esterase hydrolysis (36.1% of administered dose) and secondarily by CYP3A4-mediated oxidative (21.2% of administered dose) pathways. Other non-CYP3A4 metabolites account for 7.2% of the administered dose. Glucuronidation is a minor metabolic pathway (< 1% of administered dose).
Temsavir is extensively metabolized, accounting for the fact that only 3% of the administered dose is recovered in human urine and faeces. Temsavir is biotransformed into two predominant circulating inactive metabolites, BMS-646915 (a product of hydrolysis) and BMS-930644 (a product of N-dealkylation). Drugs that are strong inducers of CYP3A are contraindicated with fostemsavir (see Section 4.3 Contraindications; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Excretion.
Temsavir has a terminal half-life of approximately 11 hours. Plasma temsavir clearance following intravenous administration was 17.9 L/hr, and the apparent clearance (CL/F) following oral dosing was 66.4 L/hr. After oral administration of a single 300 mg dose of 14C-labeled fostemsavir in a human mass balance study, 51% and 33% of the radioactivity was retrieved in the urine and faeces, respectively. Based on limited bile collection in this study (3 to 8 hours post dose), biliary clearance accounted for 5% of the radioactive dose, suggesting that a fraction of faecal excretion is from biliary excretion.
Special patient populations.
Children.
The pharmacokinetics of temsavir have not been evaluated in children younger than 18 years.
Elderly.
Population pharmacokinetic analysis of temsavir using data in HIV-1 infected adults showed that there was no clinically relevant effect of age on temsavir exposure. Pharmacokinetic data for temsavir in subjects aged 65 years and older are limited. Elderly patients may be more susceptible to drug-induced QT interval prolongation (see Section 4.4 Special Warnings and Precautions for Use).
Renal impairment.
No dosage adjustment of fostemsavir is required for patients with mild, moderate, or severe renal impairment and for patients with end stage renal disease (ESRD). The effect of renal impairment on the exposure of temsavir after a single 600 mg dose of fostemsavir was evaluated in an open-label study in 30 adult subjects with normal renal function, mild, moderate, and severe renal impairment, and subjects with ESRD on haemodialysis (n = 6 per group). Classification of renal function was based on estimated glomerular filtration rate (eGFR), as follows: 60 ≤ eGFR ≤ 89 (mild), 30 ≤ eGFR < 60 (moderate), eGFR < 30 (severe, and ESRD on haemodialysis) mL/min/1.73 m2. There was no effect of renal impairment on total and unbound temsavir Cmax and AUC based on both eGFR and creatinine clearance (CLcr); however, the upper bounds of the 2-sided 90% CI for the impact of renal impairment on plasma unbound temsavir Cmax are as high as 2.7-fold with severe renal impairment which is lower than the 4.2-fold Cmax threshold established based on temsavir exposure-safety response relationship (see Section 5.1 Pharmacodynamic Properties, Effects on electrocardiogram). Thus, increases in temsavir exposures are not considered clinically relevant.
Fostemsavir may be administered to patients with ESRD without regard to time of haemodialysis because temsavir was not readily cleared by haemodialysis, with approximately 12.3% of the administered dose removed during the 4-hour haemodialysis session. Haemodialysis initiated 4 hours after temsavir dosing was associated with an average 46% increase in plasma total temsavir Cmax and an average 11% decrease in AUC relative to pharmacokinetics off haemodialysis.
Hepatic impairment.
No dosage adjustment of fostemsavir is necessary for patients with mild, moderate and severe hepatic impairment. The effect of hepatic impairment on the exposure of temsavir after a single 600 mg dose of fostemsavir was evaluated in an open-label study in 30 adult subjects with normal (n = 12), mild (Child-Pugh Score A, n = 6), moderate (Child-Pugh Score B, n = 6), and severe (Child-Pugh Score C, n = 6) hepatic impairment. Total and unbound temsavir exposures increased with increasing severity of hepatic impairment classified by Child-Pugh classes. In patients with mild to severe hepatic impairment, the increased exposures to both unbound and total geometric mean temsavir Cmax and AUC range from 1.2- to 2.2-fold; however, the upper bounds of the 2-sided 90% CI for the impact of hepatic impairment on plasma unbound temsavir Cmax are as high as 3-fold with severe hepatic impairment which is lower than the 4.2-fold Cmax threshold established based on temsavir exposure-safety response relationship (see Section 5.1 Pharmacodynamic Properties, Effects on electrocardiogram). Thus, increases in temsavir exposures are not considered clinically relevant.
Gender.
Population pharmacokinetic analyses indicated no clinically relevant effect of gender on the exposure of temsavir. Of the 764 subjects included in the analysis, 216 (28%) were female.
Race.
Population pharmacokinetic analyses indicated no clinically relevant effect of race on the exposure of temsavir. Of the 764 subjects included in the analysis, 490 (64%) were White, 177 (23%) were Black/African American, 5 (1%) were Asian, and 92 (12%) were of "other" race.
Co-infection with hepatitis B or C.
Pharmacokinetic data for temsavir in patients co-infected with hepatitis B and/or C virus are limited.
5.3 Preclinical Safety Data
Genotoxicity.
Neither fostemsavir nor temsavir were mutagenic or clastogenic using in vitro tests in bacteria and cultured mammalian cells and an in vivo rat micronucleus assay.
Carcinogenicity.
In a 2-year carcinogenicity study conducted in rats and a 26-week carcinogenicity study conducted in transgenic mice, fostemsavir produced no statistically significant increases in tumours over controls. The maximum daily exposures in transgenic mice were approximately 18 times (males) and 39 times (females) greater than those in humans at the maximum recommended human dose, based on AUC. The maximum daily exposures in rats were approximately 6 times (males) and 38 times (females) greater than those in humans at the maximum recommended human dose.6 Pharmaceutical Particulars
6.1 List of Excipients
Colloidal anhydrous silica; hyprolose; hypromellose; magnesium stearate; Opadry II complete film coating system 85F170022 Beige.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Fostemsavir tablets are supplied in white high-density polyethylene (HDPE) bottles with polypropylene child resistant closures that include a polyethylene faced induction heat seal liner. Each bottle contains 60 film-coated tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
Fostemsavir is a prodrug of temsavir, a human immunodeficiency virus (HIV) attachment inhibitor. The chemical name of fostemsavir trometamol is (3-((4-benzoyl-1-piperazinyl)(oxo)acetyl)-4-methoxy-7-(3-methyl-1H-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-1-yl)methyl dihydrogen phosphate, 2-amino-2-(hydroxymethyl)-1,3-propanediol (1:1). The empirical formula is C25H26N7O8P.C4H11NO3. The molecular weight is 704.6 g per mol. It has the following structural formula:
 Molecular formula: C25H26N7O8P·C4H11NO3.
Molecular formula: C25H26N7O8P·C4H11NO3.
Molecular mass: 704.62 (trometamol salt).
CAS number.
864953-39-9 (trometamol salt).7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes