1 Name of Medicine
Amivantamab.
2 Qualitative and Quantitative Composition
Each single-use vial contains 350 mg of amivantamab per 7 mL vial (or 50 mg of amivantamab per mL).
Amivantamab is a fully human immunoglobulin G1 (IgG1)-based bispecific antibody directed against the epidermal growth factor (EGF) and mesenchymal-epidermal transition (MET) receptors, produced by a mammalian cell line (Chinese Hamster Ovary [CHO]) using recombinant DNA technology (see Section 5.1, Mechanism of action).
For the full list of excipients, see Section 6.1.
3 Pharmaceutical Form
Concentrate for solution for infusion (injection).
Rybrevant is available as a colourless to pale yellow preservative-free liquid concentrate for intravenous infusion after dilution.
4.1 Therapeutic Indications
Rybrevant is indicated:
In combination with lazertinib for the first-line treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations.
In combination with carboplatin and pemetrexed for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal-growth factor receptor (EGFR) exon 19 deletions or exon 21 L858R substitution mutations, whose disease has progressed on or after treatment with an EGFR tyrosine kinase inhibitor (TKI).
In combination with carboplatin and pemetrexed for the first-line treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations.
As monotherapy for the treatment of adult patients with locally advanced or metastatic NSCLC with activating EGFR exon 20 insertion mutations whose disease has progressed on or after platinum-based chemotherapy.
4.2 Dose and Method of Administration
Rybrevant should be administered by a healthcare professional in a setting with appropriate medical support for the management of infusion-related reactions (IRRs), including equipment for cardiorespiratory resuscitation. See Section 4.4 Special Warnings and Precautions for Use.
Administer pre-infusion medications (see Pre-infusion medications).
Administer diluted Rybrevant intravenously according to the infusion rates in Table 1 and Table 2, with the initial dose as a split across two infusions - on Day 1 and Day 2 of week 1.
Prior to the use of Rybrevant, the presence of EGFR mutation exon 19 deletion, exon 21 L858R substitution, or exon 20 insertion mutation must be established (see Section 5.1, Clinical trials).
When initiating treatment with Rybrevant in combination with lazertinib, it is recommended to administer anticoagulant prophylaxis to prevent venous thromboembolic (VTE) events for the first four months of treatment. Ongoing anticoagulation beyond four months is at clinician's discretion. Anticoagulants use should align with clinical guidelines, use of Vitamin K antagonists is not recommended (see Section 4.4 Special Warnings and Precautions for Use).
Dosage.
Dosage - adults (≥ 18 years).
Due to the frequency of IRRs at the first dose, infusion into a peripheral vein should be considered in Week 1 and Week 2 to minimise drug exposure in the event of an IRR; a central line may be used subsequently. Particularly for the first dose, prepare the dilution for infusion as close as possible to the time of administration (see Administration), to allow for maximal flexibility in IRR management.
Dose regimens.
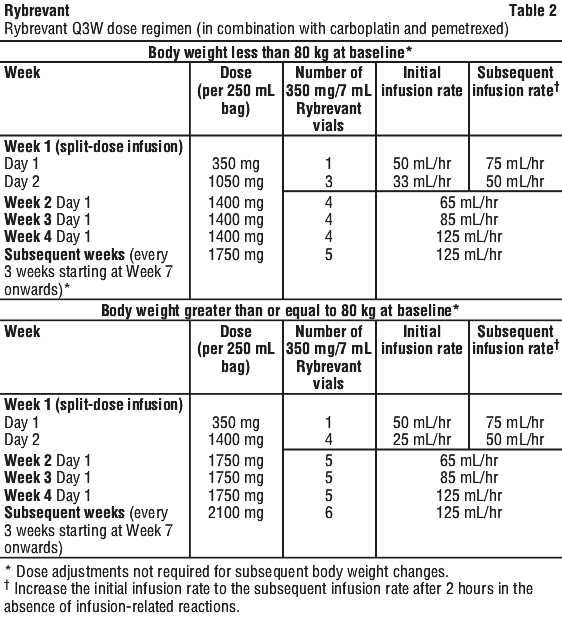
The recommended dose regimen for Rybrevant is once every two weeks (Q2W; see Table 1) when used as monotherapy or in combination with lazertinib, and once every three weeks (Q3W; see Table 2) when used in combination with carboplatin and pemetrexed (see Table 3).
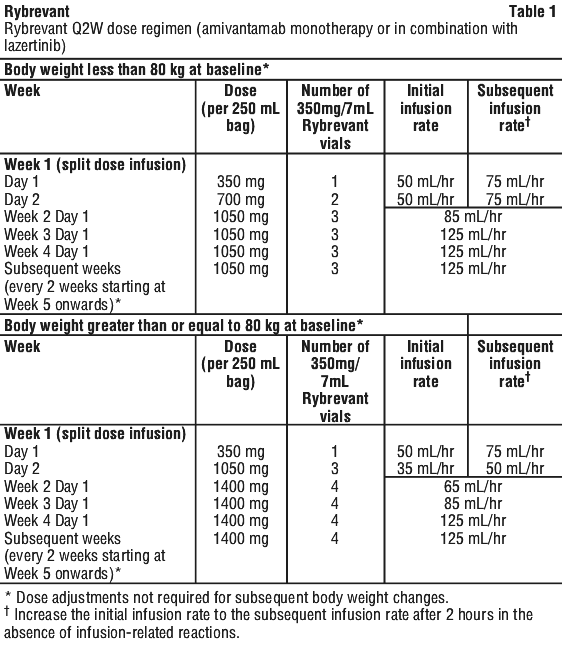 When used in combination with lazertinib, it is recommended to administer Rybrevant any time after lazertinib when given on the same day.
When used in combination with lazertinib, it is recommended to administer Rybrevant any time after lazertinib when given on the same day.
Duration of treatment.
Administer Rybrevant until disease progression or unacceptable toxicity.
Pre-infusion medications.
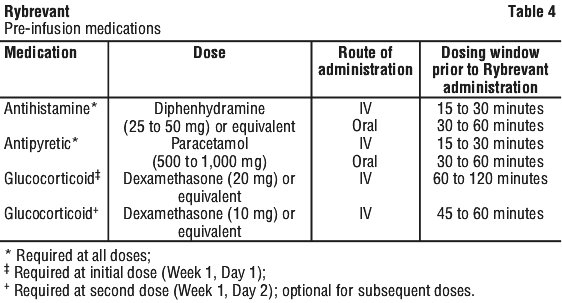
Prior to initial infusion of Rybrevant (Week 1, Days 1 and 2), administer antihistamines, antipyretics, and glucocorticoids to reduce the risk of IRRs. For subsequent doses, administer antihistamines and antipyretics. Administer antiemetics as needed. Table 4 summarises the recommendations regarding pre-infusion medications.
Missed dose(s).
If a planned dose of Rybrevant is missed, the dose should be administered as soon as possible and the dosing schedule should be adjusted accordingly, maintaining the treatment interval.
Dose modifications.
The recommended dose reductions for adverse reactions (see Table 6) are listed in Table 5.
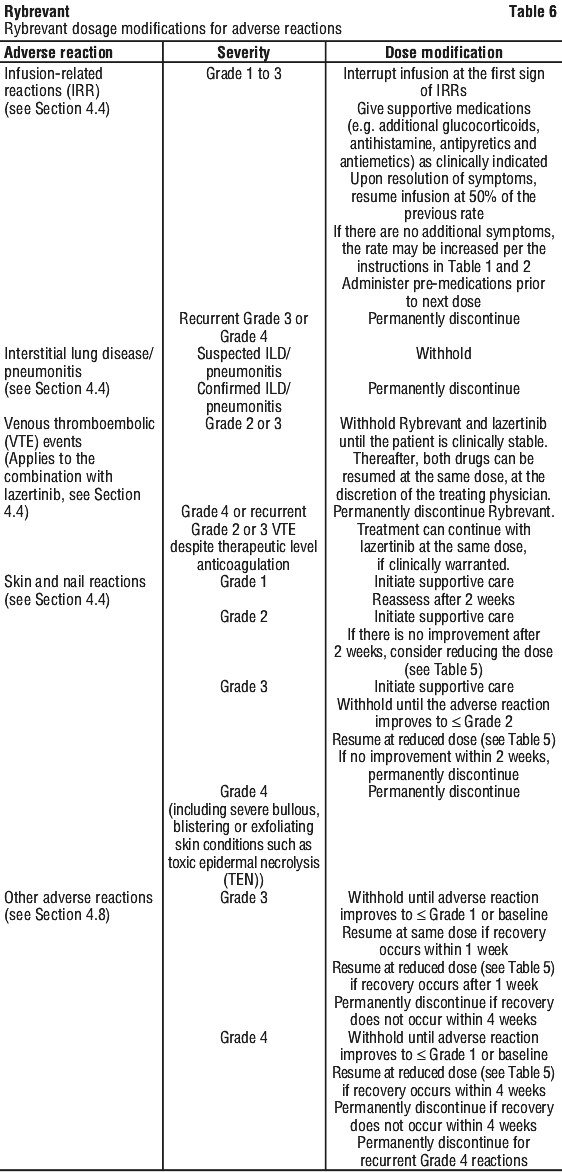
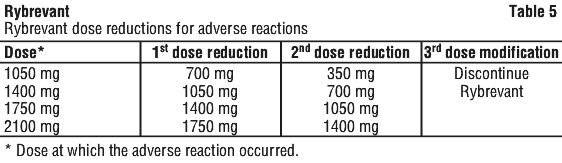 The recommended dosage modifications for adverse reactions are provided in Table 6.
The recommended dosage modifications for adverse reactions are provided in Table 6.
Special populations.
Paediatrics (17 years of age and younger).
The safety and efficacy of Rybrevant have not been established in paediatric patients.
Elderly (65 years of age and older).
Of the 661 patients treated with Rybrevant in EDI1001 (CHRYSALIS), NSC3001 (PAPILLON) and NSC3002 (MARIPOSA-2), 40% were 65 years of age or older, and 10% were 75 years of age or older. No overall differences in safety or effectiveness were observed between these patients and younger patients. No dosage adjustment is necessary (see Section 5.2 Pharmacokinetic Properties).
Renal impairment.
No formal studies of amivantamab in patients with renal impairment have been conducted. Based on population pharmacokinetic (PK) analyses, no dosage adjustment is necessary for patients with mild or moderate renal impairment. No data are available in patients with severe renal impairment (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No formal studies of amivantamab in patients with hepatic impairment have been conducted. Based on population PK analyses, no dosage adjustment is necessary for patients with mild hepatic impairment. No data are available in patients with moderate or severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Administration.
Preparation for administration.
Rybrevant solution must be diluted and prepared for intravenous infusion by a healthcare professional using aseptic technique.
1. Determine the dose required and number of Rybrevant vials needed based on patient's baseline weight (see Table 1 and Table 2). Each vial of Rybrevant contains 350 mg of amivantamab.
2. Check that the Rybrevant solution is colourless to pale yellow. Do not use if discolouration or visible particles are present.
3. Withdraw and then discard a volume of either 5% dextrose [glucose] solution or 0.9% sodium chloride solution from the 250 mL infusion bag equal to the volume of Rybrevant to be added (i.e. discard 7 mL diluent from the infusion bag for each Rybrevant vial). Infusion bags must be made of polyvinylchloride (PVC), polypropylene (PP), polyethylene (PE), or polyolefin blend (PP+PE).
4. Withdraw 7 mL of Rybrevant from each vial and add it to the infusion bag. The final volume in the infusion bag should be 250 mL. Each vial contains a 0.5 mL overfill to ensure sufficient extractable volume. Discard any unused portion left in the vial.
5. Gently invert the bag to mix the solution. Do not shake.
6. Visually inspect the diluted solution before administration. Do not use if discolouration or visible particles are observed.
7. Diluted solutions should be administered within 10 hours (including infusion time) at room temperature (15°C to 25°C) and in room light.
Administration.
1. Prior to administration, prime the infusion set with the diluent (either 5% dextrose [glucose] solution or 0.9% sodium chloride solution).
2. Administer the diluted solution by intravenous infusion using an infusion set fitted with a flow regulator and with an in-line, sterile, non-pyrogenic, low protein-binding polyethersulfone (PES) filter (pore size 0.2 micrometer). Administration sets must be made of either polyurethane (PU), polybutadiene (PBD), PVC, PP, or PE.
3. Do not infuse Rybrevant concomitantly in the same intravenous line with other agents.
4. Product is for single use in one patient only. Discard any residue.4.3 Contraindications
Hypersensitivity to amivantamab or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
The data described in Special Warnings and Precautions for Use reflects the safety profile of patients with locally advanced or metastatic NSCLC, including 380 patients who received Rybrevant monotherapy in Study EDI1001 (CHRYSALIS), 151 patients who received Rybrevant in combination with carboplatin and pemetrexed in Study NSC3001 (PAPILLON), 130 patients who received Rybrevant in combination with carboplatin and pemetrexed in Study NSC3002 (MARIPOSA-2) and 421 patients who received Rybevrant in combination with lazertinib in Study NSC3003 (MARIPOSA).
Infusion-related reactions.
IRRs may occur in patients treated with Rybrevant. The most frequent signs and symptoms include chills, nausea, dyspnoea, flushing, chest discomfort, and vomiting.
IRRs were reported in 61% of patients treated with Rybrevant, of which 93% were Grade 1-2. A majority of IRRs occurred at the first infusion with a median time to onset of 60 minutes. Signs and symptoms of IRR include dyspnoea, flushing, fever, chills, chest discomfort, hypotension, nausea and vomiting.
To reduce the risk of IRRs, premedicate with antihistamines, antipyretics, and glucocorticoids, and follow the infusion recommendations in Section 4.2 Dose and Method of Administration.
Give Rybrevant infusions in a monitored setting with appropriate medical support for the treatment of IRRs, including cardiopulmonary resuscitation medication and equipment. Interrupt infusion if IRR is suspected, and reduce infusion rate or permanently discontinue Rybrevant based on severity (see Section 4.2 Dose and Method of Administration, Table 6).
Interstitial lung disease/pneumonitis.
Interstitial lung disease (ILD)/ pneumonitis occurred in 2.7% of patients treated with Rybrevant, including 0.1% fatal events. Patients with a medical history of ILD, drug-induced ILD, radiation pneumonitis that required steroid treatment, or any evidence of clinically active ILD have not been studied.
Monitor patients for new or worsening symptoms indicative of ILD/pneumonitis (e.g. dyspnoea, cough, fever). Immediately withhold Rybrevant in patients with suspected ILD/pneumonitis and permanently discontinue if ILD/ pneumonitis is confirmed (see Section 4.2 Dose and Method of Administration, Table 6).
Venous thromboembolic (VTE) events with concomitant use with lazertinib.
In patients receiving Rybrevant in combination with lazertinib, venous thromboembolic (VTE) events, including deep venous thrombosis (DVT) and pulmonary embolism (PE) occurred in 36% of patients, predominantly in the first four months of therapy, including 0.5% fatal events. Prophylactic anticoagulants are recommended to be used for the first four months of treatment. Ongoing anticoagulation beyond four months is at clinician's discretion. Anticoagulants use should align with clinical guidelines, use of Vitamin K antagonists is not recommended.
For VTE events associated with clinical instability, Rybrevant and lazertinib should be withheld until the patient is clinically stable. Thereafter, both drugs can be resumed at the discretion of the treating physician.
In the event of recurrence despite appropriate anticoagulation, permanently discontinue Rybfrevant. Treatment can continue with lazertinib at the same dose, if clinically warranted (see Section 4.2 Dose and Method of Administration, Table 6).
Skin and nail reactions.
Skin and nail reactions may occur in patients treated with Rybrevant.
Rash (including dermatitis acneiform), pruritus and dry skin occurred in patients treated with Rybrevant. Most cases were Grade 1 or 2, with Grade 3 events occurring in 15.5% of patients. Rash leading to Rybrevant discontinuation occurred in 2.9% of patients. Rash usually developed within the first 4 weeks of therapy, with a median time to onset of 14 days. Nail toxicity occurred in patients treated with Rybrevant. Most events were Grade 1 or 2, with Grade 3 nail toxicity occurring in 6.3% of patients.
Toxic epidermal necrolysis (TEN) has been reported. Permanently discontinue Rybrevant if TEN is confirmed.
A prophylactic approach to rash prevention should be considered. Instruct patients to limit sun exposure during and for 2 months after Rybrevant therapy. Protective clothing and use of sunscreen is advisable. Alcohol-free emollient cream is recommended for dry skin. If skin or nail reactions develop, start topical corticosteroids and topical and/or oral antibiotics. For Grade 3 or poorly-tolerated Grade 2 events, add systemic antibiotics and oral steroids and consider dermatology consultation. Promptly refer patients presenting with severe rash, atypical appearance or distribution, or lack of improvement within 2 weeks to a dermatologist. Withhold, dose reduce, or permanently discontinue Rybrevant based on severity (see Section 4.2 Dose and Method of Administration, Table 6).
Eye disorders.
Eye disorders, including keratitis (1.3%) and uveitis (0.09%) occurred in patients treated with Rybrevant. Other reported adverse reactions included dry eye, blurred vision, eye pruritus, visual impairment, aberrant eyelash growth, ocular hyperaemia, conjunctival hyperaemia and blepharitis. Most events were Grade 1-2. Refer patients presenting with worsening eye symptoms promptly to an ophthalmologist and advise discontinuation of contact lenses until symptoms are evaluated. Withhold, dose reduce, or permanently discontinue Rybrevant based on severity (see Section 4.2 Dose and Method of Administration, Table 6).
Use in the elderly.
See Section 4.2 Dose and Method of Administration.
Paediatric use.
The safety and efficacy of Rybrevant have not been established in paediatric patients.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No drug interaction studies have been performed.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
The effect of amivantamab on fertility has not been studied.
(Category D)
Based on its mechanism of action and findings in animal studies, amivantamab could cause fetal harm if administered to a pregnant patient. Whilst the use of amivantamab during pregnancy has not been studied, administration of other EGFR or MET inhibitor molecules to pregnant animals has resulted in an increased incidence of impairment of embryo-fetal development, embryolethality, and abortion. Human IgG1 is known to cross the placenta; therefore, amivantamab has the potential to be transmitted from a pregnant patient to the developing fetus. Advise patients of the potential risk to a fetus. Advise female patients of reproductive potential to use effective contraception (such as condoms) during treatment and for 3 months after the last dose of Rybrevant. Advise male patients not to donate or store semen and to use effective contraception during treatment and for 3 months after the last dose of Rybrevant.
It is not known whether amivantamab is excreted in milk or affects milk production. Because of the potential for serious adverse reactions from Rybrevant in a breastfed child, advise patients not to breastfeed during treatment with Rybrevant and for 3 months following the last dose of Rybrevant.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed. If patients experience treatment-related symptoms affecting their ability to concentrate and react, it is recommended that they do not drive or use machines whilst affected.
4.8 Adverse Effects (Undesirable Effects)
Clinical trial data.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
First-line treatment of NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutation.
The safety data described below reflect exposure to Rybrevant in combination with lazertinib in 421 treatment-naïve patients with locally advanced or metastatic NSCLC whose tumours have EGFR exon 19 deletion or exon 21 L858R substitution mutation in MARIPOSA.
Patients received lazertinib 240 mg orally once daily and Rybrevant intravenously at 1050 mg (for patients < 80 kg) or 1400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5. Median treatment duration was 18.5 months (range: 0.2 to 31.4 months) for the Rybrevant in combination with lazertinib arm and the median treatment duration was 18.00 months (range: 0.2 to 32.7 months) for the osimertinib arm. Among the 421 patients who received Rybrevant in combination with lazertinib, 73% were exposed to Rybrevant for ≥ 6 months and 60% were exposed to Rybrevant for > 1 year.
Serious adverse reactions occurred in 49% of patients who received Rybrevant in combination with lazertinib. Serious adverse reactions occurring in ≥ 2% of patients included VTE (11%), pneumonia (4%), rash, and ILD/pneumonitis (2.9% each), COVID-19 (2.4%), pleural effusion and IRR (2.1% each). Fatal adverse reactions occurred in 7% of patients who received Rybrevant in combination with lazertinib due to death not otherwise specified (1.2%); sepsis and respiratory failure (1% each); pneumonia, myocardial infarction and sudden death (0.7% each); cerebral infarction, pulmonary embolism (PE), and COVID-19 infection (0.5% each); and ILD/pneumonitis, acute respiratory distress syndrome (ARDS), and cardiopulmonary arrest (0.2% each).
Permanent discontinuation of Rybrevant due to an adverse reaction occurred in 34% of patients. Adverse reactions which resulted in permanent discontinuation in ≥ 1% of patients included rash, IRR, nail toxicity, VTE, ILD/pneumonitis, pneumonia, oedema, hypoalbuminaemia, fatigue, paraesthesia and dyspnoea.
Dose interruptions of Rybrevant due to an adverse reaction occurred in 88% of patients. Adverse reactions which required dosage interruption in ≥ 5% of patients were IRRs, rash, nail toxicity, COVID-19, VTE, increased ALT, oedema, and hypoalbuminaemia.
Dose reductions of Rybrevant due to an adverse reaction occurred in 46% of patients. Adverse reactions requiring dose reductions in ≥ 5% of patients were rash and nail toxicity.
The most common adverse reactions (≥ 20%) were rash, nail toxicity, IRR, musculoskeletal pain, stomatitis, oedema, VTE, paraesthesia, fatigue, diarrhoea, constipation, COVID-19, haemorrhage, dry skin, decreased appetite, pruritus, and nausea. The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were decreased albumin, decreased sodium, increased alanine aminotransferase, decreased potassium, decreased haemoglobin, increased aspartate aminotransferase, increased gamma glutamyl transferase, and increased magnesium.
Table 7 summarises the adverse reactions (≥ 10%) in MARIPOSA.
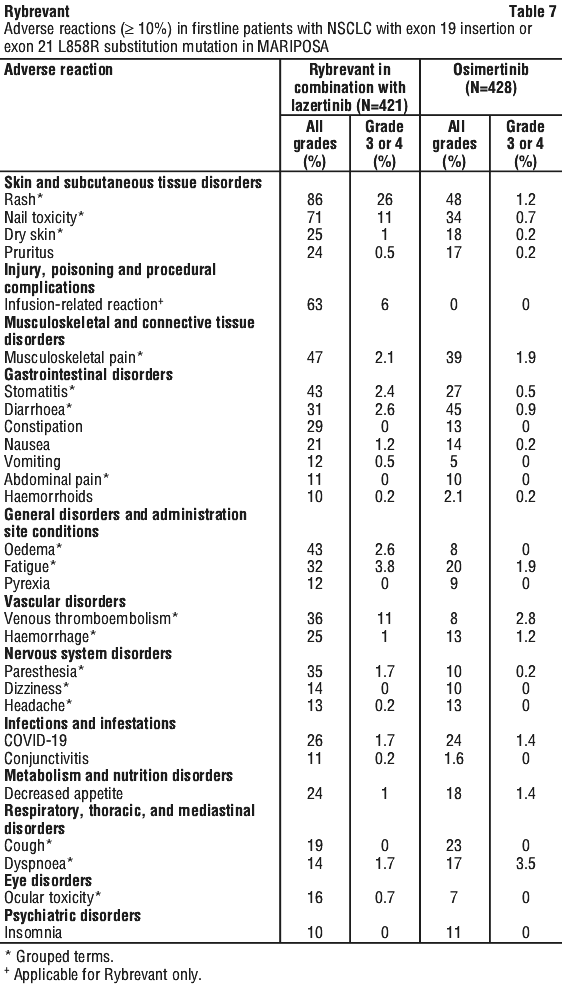 Clinically relevant adverse reactions in < 10% of patients who received Rybrevant in combination with lazertinib included ILD/pneumonitis (3.1%).
Clinically relevant adverse reactions in < 10% of patients who received Rybrevant in combination with lazertinib included ILD/pneumonitis (3.1%).
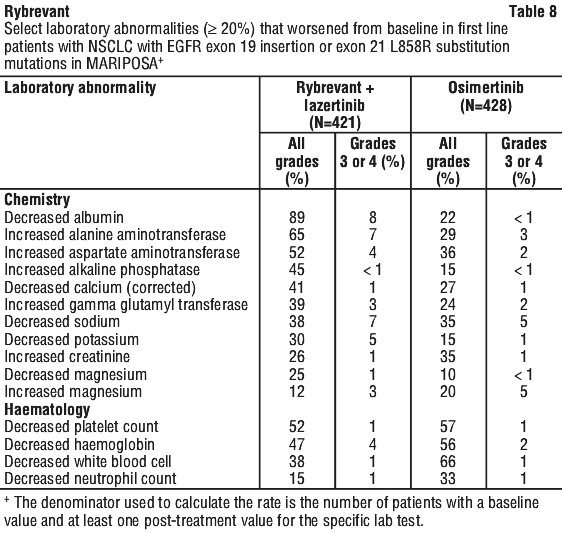
Laboratory abnormalities.
Table 8 summarises the laboratory abnormalities in MARIPOSA.
Treatment of NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations after prior therapy.
The safety of Rybrevant in combination with carboplatin and pemetrexed at the recommended dosage (see Table 1 and 2) was evaluated in the MARIPOSA-2 study (see Section 5.1 Pharmacodynamic Properties, Clinical trials), conducted in patients with locally advanced or metastatic NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations whose disease had progressed on or after treatment with osimertinib. The median (range) treatment duration was 6.3 (0 to 14.7) months amongst 130 patients who received the combination including Rybrevant, and 3.7 (0 to 15.9) months amongst 243 patients who received carboplatin plus pemetrexed in the comparator arm.
The median age was 62 years (range: 36 to 84 years); 62% were female; 48% were Asian, and 46% were White; and 87% had baseline body weight < 80 kg.
Serious adverse reactions occurred in 32% of patients who received Rybrevant in combination with carboplatin and pemetrexed. Serious adverse reactions that were reported in > 2% of patients included dyspnoea (3.1%), thrombocytopenia (3.1%), sepsis (2.3%), and pulmonary embolism (2.3%). Fatal adverse reactions occurred in 2.3% of patients who received Rybrevant in combination with carboplatin and pemetrexed; these included respiratory failure, sepsis, and ventricular fibrillation (0.8% each).
Dose interruptions of Rybrevant due to an adverse reaction occurred in 60% of patients. IRR requiring infusion interruptions occurred in 52% of patients. Adverse reactions requiring dose interruption in ≥ 5% of patients included infusion-related reactions, rash and fatigue.
Dose reductions of Rybrevant due to an adverse reaction occurred in 17% of patients. Adverse reactions requiring dose reductions in ≥ 2% of patients included rash.
Eleven percent of patients permanently discontinued Rybrevant due to adverse reactions. The most frequent adverse reactions that led to treatment discontinuation (≥ 5% of patients) were IRRs.
Table 9 summarises the most common adverse reactions in MARIPOSA-2.
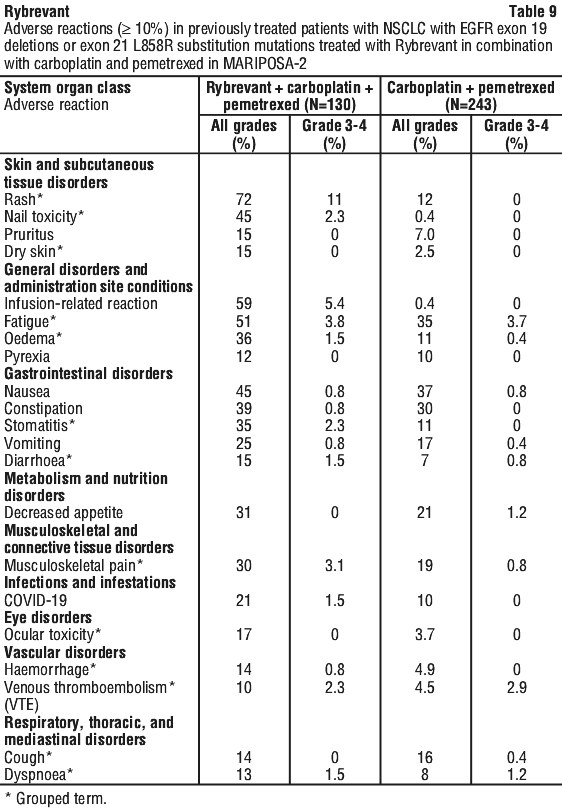 Clinically relevant adverse reactions in < 10% of patients who received Rybrevant in combination with carboplatin and pemetrexed include: abdominal pain, haemorrhoids, dizziness, visual impairment, trichomegaly, keratitis, and interstitial lung disease.
Clinically relevant adverse reactions in < 10% of patients who received Rybrevant in combination with carboplatin and pemetrexed include: abdominal pain, haemorrhoids, dizziness, visual impairment, trichomegaly, keratitis, and interstitial lung disease.
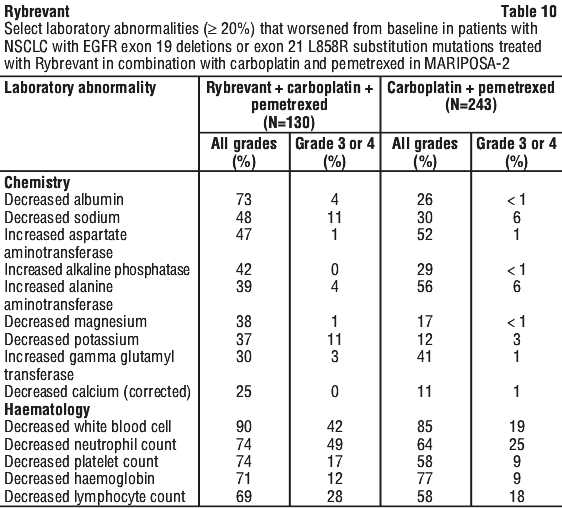
Table 10 summarises the laboratory abnormalities in MARIPOSA-2.
First-line treatment of NSCLC with exon 20 insertion mutations.
The safety of Rybrevant in combination with carboplatin and pemetrexed at the recommended dosage (see Table 1 and 2) was evaluated in the PAPILLON study (see Section 5.1 Pharmacodynamic Properties, Clinical trials) conducted in patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Among 151 patients who received Rybrevant in combination with carboplatin and pemetrexed the median exposure was 9.7 months (range: 0.0 to 26.9 months).
The median age was 61 years (range: 27 to 86 years); 56% were female; 64% were Asian, 32% were White and 86% had baseline body weight < 80 kg.
Serious adverse reactions occurred in 37% of patients who received Rybrevant in combination with carboplatin and pemetrexed. Serious adverse reactions in ≥ 2% of patients included rash, pneumonia, interstitial lung disease, pulmonary embolism, vomiting, and COVID-19. Fatal adverse reactions occurred in 7 patients (4.6%) due to pneumonia, cerebrovascular accident, cardiorespiratory arrest, COVID-19, sepsis, and death not otherwise specified.
Permanent discontinuation of Rybrevant due to an adverse reaction occurred in 11% of patients. Adverse reactions resulting in permanent discontinuation of Rybrevant in ≥ 1% of patients were rash and ILD.
Dose interruptions of Rybrevant due to an adverse reaction occurred in 64% of patients. IRR requiring infusion interruptions occurred in 38% of patients. Adverse reactions requiring dose interruption in ≥ 5% of patients included rash, nail toxicity, and hypokalaemia.
Dose reductions of Rybrevant due to an adverse reaction occurred in 36% of patients. Adverse reactions requiring dose reductions in ≥ 5% of patients included rash, and nail toxicity.
Table 11 summarises the most common adverse reactions in PAPILLON.
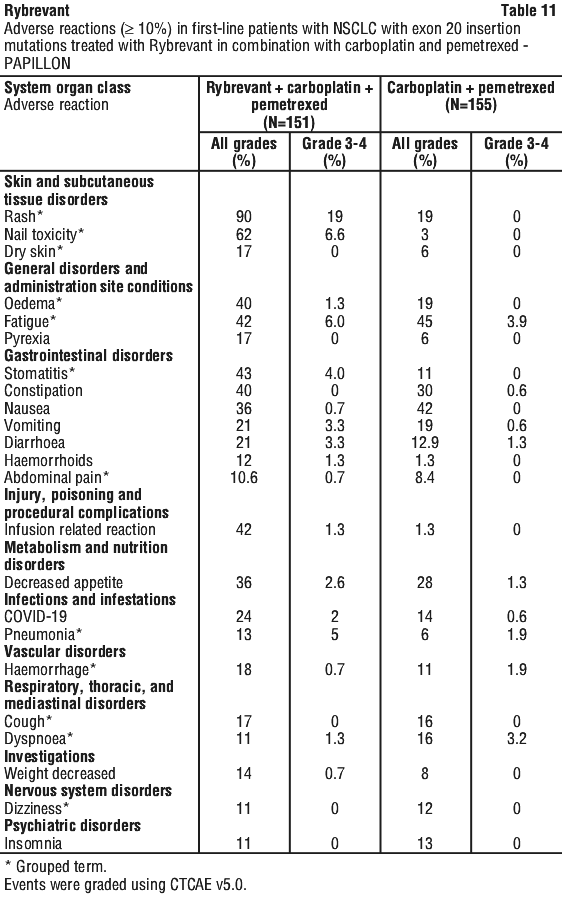 Clinically relevant adverse reactions in < 10% of patients who received Rybrevant in combination with carboplatin and pemetrexed included pulmonary embolism, deep vein thrombosis, skin ulcer, conjunctivitis, and interstitial lung disease (ILD)/ pneumonitis.
Clinically relevant adverse reactions in < 10% of patients who received Rybrevant in combination with carboplatin and pemetrexed included pulmonary embolism, deep vein thrombosis, skin ulcer, conjunctivitis, and interstitial lung disease (ILD)/ pneumonitis.
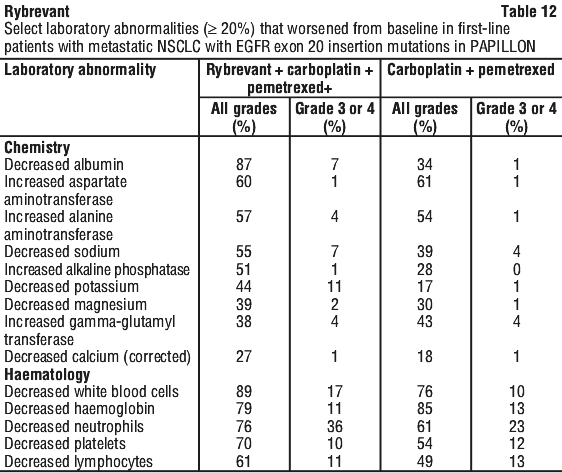
Table 12 summarises the laboratory abnormalities in PAPILLON.
Treatment of NSCLC with EGFR exon 20 insertion mutations after prior therapy.
The safety of Rybrevant as monotherapy at the recommended dosage (see Table 1) was evaluated in CHRYSALIS, which included 153 patients with locally advanced or metastatic NSCLC with EGFR exon 20 mutations whose disease had progressed on or after platinum-based chemotherapy (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Patients received Rybrevant 1050 mg (for patients < 80 kg) or 1400 mg (for patients ≥ 80 kg) by intravenous infusion once weekly for 4 weeks, then every 2 weeks starting at Week 5 until disease progression or unacceptable toxicity. The median treatment duration was 5.6 months (range: 0.0 to 23.9 months): 46% of patients were exposed for 6 months or longer and 22% were exposed for longer than a year.
The most common adverse reactions (≥ 20% incidence) were rash, IRR, nail toxicity, hypoalbuminaemia, fatigue, oedema, stomatitis, nausea, constipation, dry skin, and alanine aminotransferase increased. Serious adverse reactions in > 1% of patients included ILD, diarrhoea, IRR, and rash. Five percent of patients discontinued Rybrevant due to adverse reactions. The most frequent adverse reactions leading to treatment discontinuation were IRR and ILD. See Table 13.
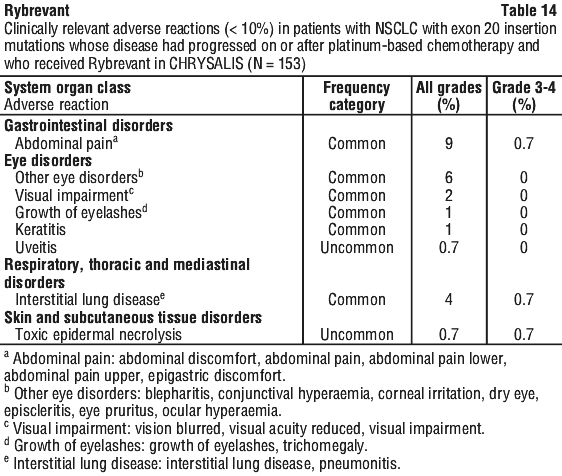
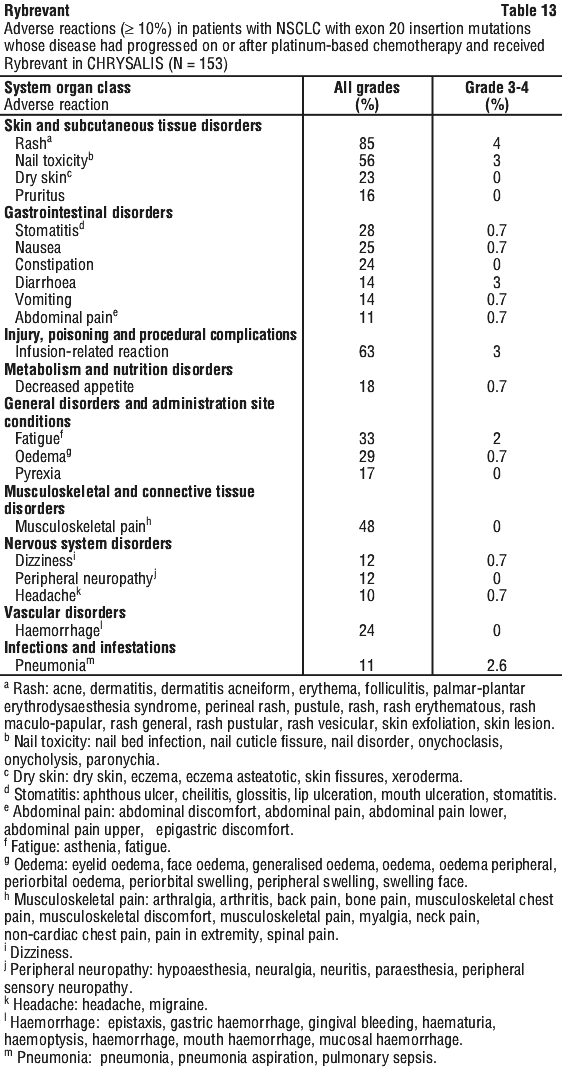 Clinically relevant adverse reactions that occurred in < 10% of Rybrevant-treated patients with NSCLC exon 20 insertion mutations in CHRYSALIS included those summarised in Table 14.
Clinically relevant adverse reactions that occurred in < 10% of Rybrevant-treated patients with NSCLC exon 20 insertion mutations in CHRYSALIS included those summarised in Table 14.
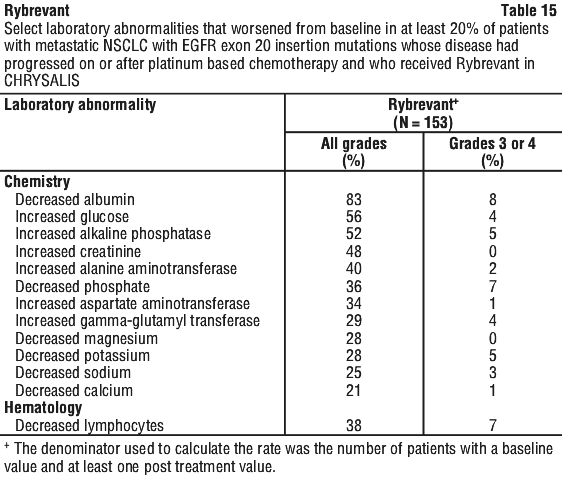
Laboratory abnormalities.
See Table 15.
Postmarketing experience.
The following adverse reactions associated with the use of Rybrevant were identified in clinical studies or postmarketing reports. Because some of these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and subcutaneous tissue disorders.
Skin ulcer.
Blood and lymphatic system disorders.
Thrombocytopenia*, neutropenia* (*only applicable to Rybrevant in combination with carboplatin and pemetrexed).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no information on overdosage with Rybrevant.
There is no known specific antidote for Rybrevant overdose. In the event of an overdose, stop Rybrevant, undertake general supportive measures until clinical toxicity has diminished or resolved.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Monoclonal antibodies and antibody drug conjugates, ATC code: L01FX18.
Mechanism of action.
Amivantamab is a low-fucose, fully-human IgG1-based bispecific antibody that binds to the extracellular domains of EGFR and MET.
In preclinical studies, amivantamab disrupted EGFR and MET signalling functions through blocking ligand binding and, in exon 20 insertion mutation models, enhancing degradation of EGFR and MET. The presence of EGFR and MET on the surface of tumour cells also allows for targeting of these cells for destruction by immune effector cells, such as natural killer cells and macrophages, through antibody-dependent cellular cytotoxicity (ADCC) and trogocytosis mechanisms, respectively.
Pharmacodynamic effects.
Immunogenicity.
Monoclonal antibodies can be immunogenic. Across the MARIPOSA, MARIPOSA-2, PAPILLON and CHRYSALIS clinical studies, there were a total of 1862 patients who received Rybrevant (either as monotherapy or as part of a combination therapy) and had evaluable results for anti-drug antibody (ADA) testing. In this group, the incidence of treatment-emergent anti-amivantamab antibodies was 0.2% (n=4). Due to the small incidence, the effect of these antibodies on the pharmacokinetics, safety or efficacy of Rybrevant can't be meaningfully assessed.
Clinical trials.
NSCLC patients with EGFR exon 19 deletions or exon 21 L858R substitution mutations previously untreated.
MARIPOSA is a randomised, active-controlled, multicentre phase 3 study assessing the efficacy and safety of Rybrevant in combination with lazertinib as compared to osimertinib monotherapy as first-line treatment in patients with EGFR-mutated locally advanced or metastatic NSCLC not amenable to curative therapy. Patient samples were required to have one of the two common EGFR mutations (exon 19 deletion or exon 21 L858R substitution mutation), as identified by local testing.
A total of 1074 patients were randomised (2:2:1) to receive Rybrevant in combination with lazertinib, osimertinib monotherapy, or lazertinib monotherapy (an unapproved regimen for NSCLC) until disease progression or unacceptable toxicity. Rybrevant was administered intravenously at 1050 mg (for patients < 80 kg) or 1400 mg (for patients ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5. Lazertinib was administered at 240 mg orally once daily. Osimertinib was administered at a dose of 80 mg orally once daily. Randomisation was stratified by EGFR mutation type (exon 19 deletion or exon 21 L858R), race (Asian or non-Asian), and history of brain metastasis (yes or no).
Baseline demographics and disease characteristics were balanced across the treatment arms. The median age was 63 (range: 25-88) years with 45% of patients ≥ 65 years; 62% were female; and 59% were Asian, and 38% were White. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (34%) or 1 (66%); 69% never smoked; 41% had prior brain metastases; and 90% had Stage IV cancer at initial diagnosis. With regard to EGFR mutation status, 60% were exon 19 deletions and 40% were exon 21 L858R substitution mutations.
Rybrevant in combination with lazertinib demonstrated a statistically significant and clinically meaningful improvement in progression-free survival (PFS) by BICR assessment, with a 30% reduction in the risk of progression or death compared with osimertinib (HR=0.70 [95% CI: 0.58, 0.85], p=0.0002). The corresponding median PFS was 23.72 months (95% CI: 19.12, 27.66) for the Rybrevant in combination with lazertinib arm and 16.59 months (95% CI: 14.78, 18.46) for the osimertinib arm.
While OS results were immature at the current analysis, with 55% of pre-specified deaths for the final analysis reported, no trend towards a detriment was observed. See Table 16 and Figure 1.
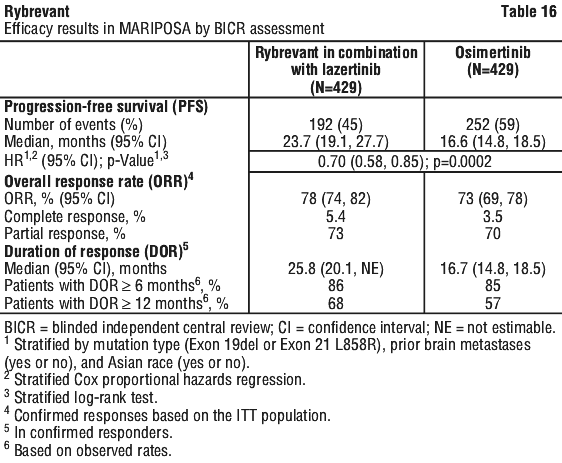
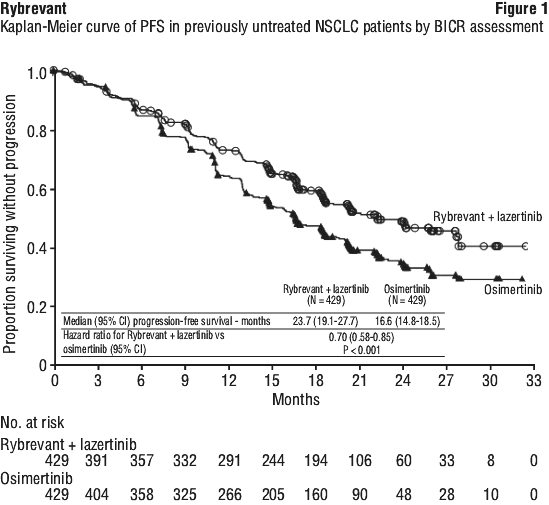 The PFS benefit of Rybrevant in combination with lazertinib as compared to osimertinib was generally consistent across prespecified, clinically relevant subgroups, including age group, sex, race, weight, mutation type, ECOG performance status, history of smoking, and history of brain metastasis at study entry (see Figure 2).
The PFS benefit of Rybrevant in combination with lazertinib as compared to osimertinib was generally consistent across prespecified, clinically relevant subgroups, including age group, sex, race, weight, mutation type, ECOG performance status, history of smoking, and history of brain metastasis at study entry (see Figure 2).
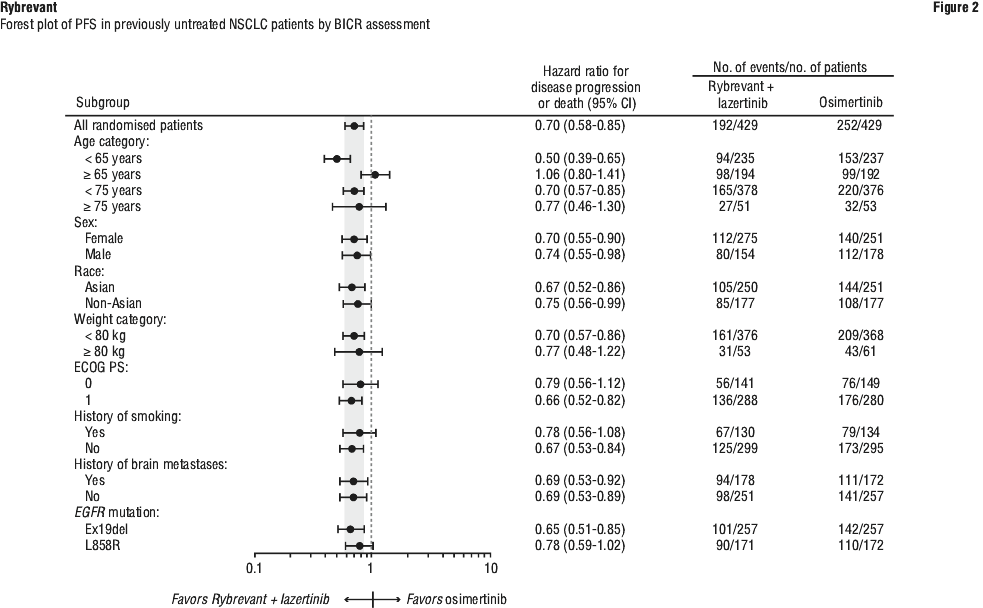 The stratified analysis of investigator-assessed PFS shows that the improved treatment effect of the combination of Rybrevant and lazertinib relative to osimertinib was also observed (median PFS of 23.92 months in the Rybrevant in combination with lazertinib arm, compared to median of 19.94 months in the osimertinib arm (HR of 0.79 [95% CI: 0.65, 0.95, nominal p=0.0139])) when assessed by investigator. Results for the analysis of ORR based on investigator assessment for comparison of the Rybrevant in combination with lazertinib arm versus the osimertinib arm were consistent with results for ORR based on BICR assessment.
The stratified analysis of investigator-assessed PFS shows that the improved treatment effect of the combination of Rybrevant and lazertinib relative to osimertinib was also observed (median PFS of 23.92 months in the Rybrevant in combination with lazertinib arm, compared to median of 19.94 months in the osimertinib arm (HR of 0.79 [95% CI: 0.65, 0.95, nominal p=0.0139])) when assessed by investigator. Results for the analysis of ORR based on investigator assessment for comparison of the Rybrevant in combination with lazertinib arm versus the osimertinib arm were consistent with results for ORR based on BICR assessment.
The MARIPOSA study included protocol-mandated brain magnetic resonance imaging (MRIs), which have historically not been used in trials evaluating EGFR-mutated NSCLC. This may have led to earlier detection of recurrences and associated shorter median values for PFS. To account for this, a sensitivity analysis was done whereby patients with brain-only progression as the site of first progression were censored. Extracranial PFS based on BICR assessment was consistent with the treatment benefit observed in the primary analysis. The median extracranial PFS was 27.5 months with Rybrevant in combination with lazertinib, as compared to 18.37 months with osimertinib (HR=0.68 [95% CI: 0.55, 0.83], nominal p=0.0001).
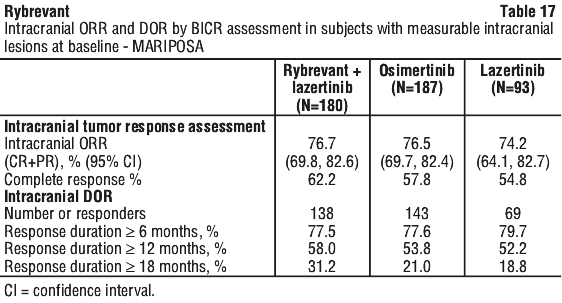
Results of pre-specified exploratory analyses of CNS ORR and DOR by BICR, in the subset of patients with measurable intracranial lesions at baseline for the combination of Rybrevant and lazertinib, demonstrated similar intracranial ORR to the control. Per protocol, all patients in MARIPOSA had serial brain MRIs to assess intracranial response and duration. Results are summarised in Table 17.
Treatment of NSCLC with EGFR exon 19 deletions or exon 21 L858R substitution mutations after prior therapy.
The efficacy of Rybrevant in combination with carboplatin and pemetrexed for advanced NSCLC harbouring a common EGFR mutation after prior anti-EGFR tyrosine kinase inhibitor therapy was evaluated in MARIPOSA-2: a randomised, open-label, multicenter trial. Eligible patients had locally advanced or metastatic NSCLC with an EGFR exon 19 deletion or exon 21 L858R substitution mutation prospectively identified by validated local testing, and had developed progressive disease on or after receiving osimertinib. Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to enrol.
A total of 131 patients were randomised to receive Rybrevant in combination with carboplatin and pemetrexed (ACP), and 263 were randomised to receive carboplatin and pemetrexed (CP). Randomisation was stratified by osimertinib line of therapy (first-line or second-line), prior brain metastases (yes or no), and Asian race (yes or no).
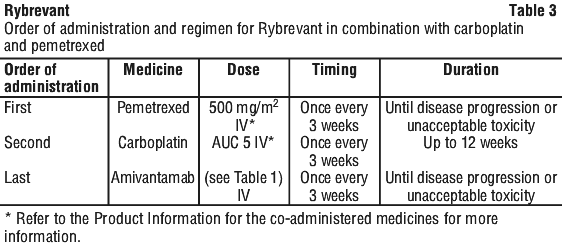
All patients received carboplatin intravenously at area under the concentration-time curve 5 mg/mL per minute (AUC 5) once every 3 weeks for up to 12 weeks. All patients also received pemetrexed intravenously at 500 mg/m2 once every 3 weeks until disease progression or unacceptable toxicity. In addition, patients randomised to the ACP arm received Rybrevant intravenously in the following regimen:
Weeks 1-4: 1400 mg (for patients < 80 kg) or 1750 mg (for patients ≥ 80 kg) per week.
Week 7 onwards: 1750 mg (for patients < 80 kg) or 2,100 mg (for patients ≥ 80 kg) once every three weeks, until disease progression or unacceptable toxicity.
Tumour assessments were performed every 6 weeks for the first 12 months and every 12 weeks thereafter. The major efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Overall survival (OS) and objective response rate (ORR) as assessed by BICR were key secondary outcome measures.
Of the 394 patients randomised to the Rybrevant-CP arm or CP arm, the median age was 62 (range: 31-85) years, and 38% were at least 65 years of age; 60% were female, 48% were Asian and 46% were Caucasian. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (40%) or 1 (60%); 66% had never smoked; 45% had a history of brain metastasis, and 92% had Stage IV cancer at initial diagnosis.
The efficacy results of MARIPOSA-2 are summarised in Table 18 and Figure 3.

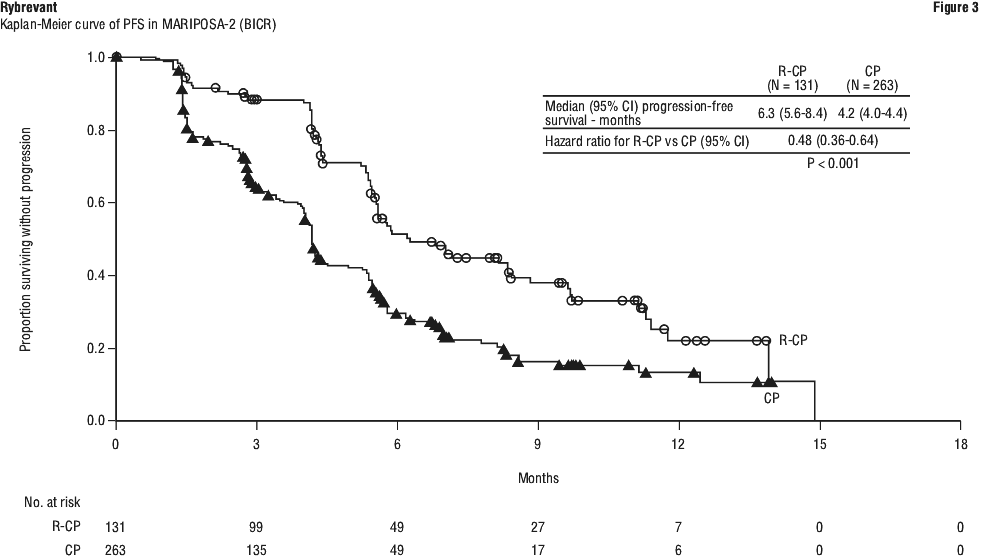 At the prespecified second interim analysis of OS, with 85% of the deaths needed for the final analysis, there was no statistically significant difference in OS. The median OS was 17.7 months (95% CI: 16.0, 22.4) in the ACP arm and 15.3 months (95% CI: 13.7, 16.8) in the CP arm, with a hazard ratio of 0.73 (95% CI: 0.54, 0.99).
At the prespecified second interim analysis of OS, with 85% of the deaths needed for the final analysis, there was no statistically significant difference in OS. The median OS was 17.7 months (95% CI: 16.0, 22.4) in the ACP arm and 15.3 months (95% CI: 13.7, 16.8) in the CP arm, with a hazard ratio of 0.73 (95% CI: 0.54, 0.99).
Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to be randomised in MARIPOSA-2. Baseline disease assessment, including brain magnetic resonance imaging (MRI) was performed at treatment initiation. All patients underwent serial brain MRI during the trial.
Pre-specified secondary analyses of intracranial ORR by BICR in the subset of 91 (23%) patients with baseline intracranial disease were performed. Data were only available for intracranial complete responses and not available for intracranial partial responses. Intracranial ORR was 20% (95% CI: 8, 39) in the 30 patients with baseline intracranial disease in the ACP arm and 7% (95% CI: 1.8, 16) in the 61 patients with baseline intracranial disease in the CP arm.
First-line treatment of locally advanced or metastatic NSCLC with exon 20 insertion mutations.
The efficacy of Rybrevant in combination with carboplatin and pemetrexed as a first line treatment for advanced NSCLC harbouring an EGFR exon 20 insertion mutation was evaluated in PAPILLON: a randomised, open-label, multicenter trial. Eligible patients had treatment-naïve, locally advanced or metastatic NSCLC with an EGFR exon 20 insertion mutation prospectively identified by validated local testing of either tumour tissue (92%) or plasma/ctDNA (8%). Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to enrol. Patients with historical or active interstitial lung disease were excluded from the clinical study.
A total of 308 subjects were randomised (1:1) to Rybrevant in combination with carboplatin and pemetrexed (N=153) or carboplatin and pemetrexed (N=155). The dosing of all agents was the same as descried above for MARIPOSA-2. Randomisation was stratified by ECOG performance status (0 vs. 1) and prior brain metastases (yes vs. no), and prior EGFR TKI use (yes vs. no).
The primary efficacy outcome measure was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Additional efficacy outcome measures included objective response rate (ORR), duration of response (DOR) and overall survival (OS). Cross-over to single agent Rybrevant was permitted for patients who had confirmed disease progression on carboplatin and pemetrexed.
The median age was 62 (range: 27 to 92) years, with 40% of the subjects ≥ 65 years of age; 58% were female; 61% were Asian and 36% were Caucasian. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (35%) or 1 (65%); 58% had never smoked; 23% had history of brain metastasis and 84% had Stage IV cancer at initial diagnosis. Four patients (including 1 in the ACP arm) had previously received an EGFR TKI, and this was omitted from stratification of analyses.
Efficacy results for PAPILLON are summarised in Table 19 and Figure 4.

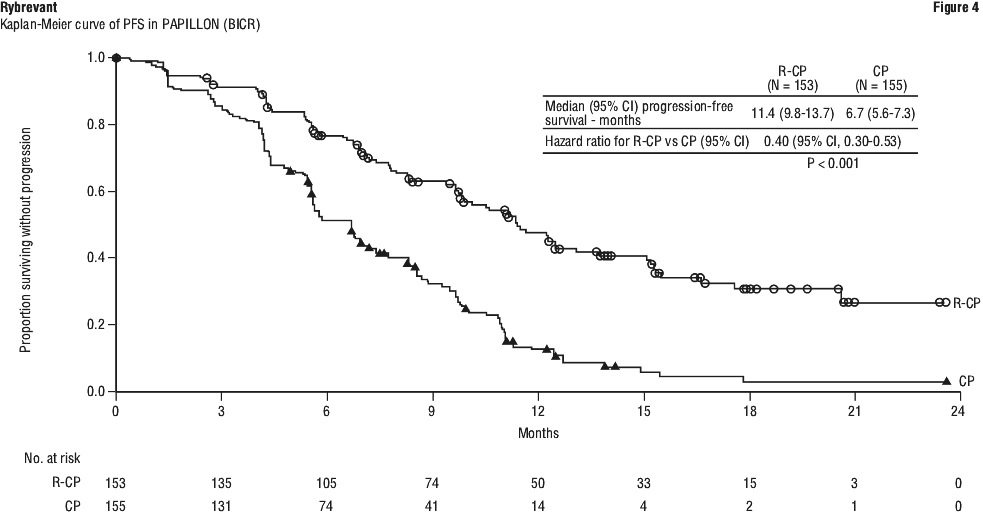 While OS results were immature at the current analysis, with 44% of pre-specified deaths for the final analysis reported, no trend towards a detriment was observed. Seventy-five (48%) of the treated patients crossed over from the carboplatin and pemetrexed arm after confirmation of disease progression to receive Rybrevant as a single agent.
While OS results were immature at the current analysis, with 44% of pre-specified deaths for the final analysis reported, no trend towards a detriment was observed. Seventy-five (48%) of the treated patients crossed over from the carboplatin and pemetrexed arm after confirmation of disease progression to receive Rybrevant as a single agent.
Treatment of locally advanced or metastatic NSCLC with exon 20 insertion mutations after prior therapy.
The efficacy and safety of Rybrevant were evaluated in CHRYSALIS: a multicentre, open-label, multi-cohort study conducted in patients with locally advanced or metastatic NSCLC. Efficacy was evaluated in 81 subjects with locally advanced or metastatic NSCLC who had EGFR exon 20 insertion mutations, whose disease had progressed on or after platinum-based chemotherapy, and who had a median follow-up of 14.5 months. Identification of an EGFR exon 20 insertion mutation was determined by prospective local testing using tumour tissue (94%) or plasma (6%) samples. Patients with untreated brain metastases and patients with a history of ILD requiring treatment with prolonged steroids or other immunosuppressive agents within the last 2 years were not eligible for the study.
Rybrevant was administered intravenously at a dose of 1050 mg (for patients < 80 kg) or 1400 mg (for subjects ≥ 80 kg) once weekly for 4 weeks, then every 2 weeks starting at Week 5 until disease progression or unacceptable toxicity. The primary efficacy endpoint was overall response rate (ORR), defined as confirmed complete response (CR) or partial response (PR) based on RECIST v1.1, assessed by a blinded independent central review (BICR). Secondary efficacy endpoints included duration of response (DOR) according to BICR.
The median age in the efficacy population was 62 (range: 42-84) years, with 9% of the patients ≥ 75 years of age; 59% were female; 49% were Asian and 37% were Caucasian; 74% had baseline body weight < 80 kg; 95% had adenocarcinoma; and 46% had received prior immunotherapy. The median number of prior therapies was 2 (range: 1 to 7). At baseline, 32% had Eastern Cooperative Oncology Group (ECOG) performance status of 0 and 67% had ECOG performance status of 1; 53% had never smoked; all patients had metastatic disease; and 22% had previous treatment for brain metastases.
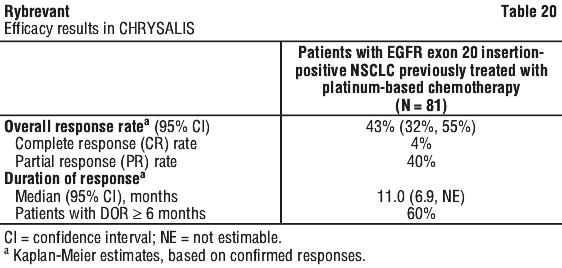
Efficacy results in the CHRYSALIS study are summarised in Table 20.
 Of the 81 patients with EGFR exon 20 insertion mutations according to local testing, plasma samples from 96% of patients were tested retrospectively using Guardant360 CDx (3.7% did not have plasma samples for testing). EGFR exon 20 insertion mutations were not identified on the Guardant360 CDx test for 20% of patients. For the 76% of patients in whom exon 20 insertion mutations were identified, the variants identified were A767 (23%), S768 (16%), D770 (11%), N771 (11%), H773 (9%), P772 (3%), V769 (1%) and A763 (1%). There were no mutation variants identified amongst the efficacy population that were associated with an absence of confirmed responses.
Of the 81 patients with EGFR exon 20 insertion mutations according to local testing, plasma samples from 96% of patients were tested retrospectively using Guardant360 CDx (3.7% did not have plasma samples for testing). EGFR exon 20 insertion mutations were not identified on the Guardant360 CDx test for 20% of patients. For the 76% of patients in whom exon 20 insertion mutations were identified, the variants identified were A767 (23%), S768 (16%), D770 (11%), N771 (11%), H773 (9%), P772 (3%), V769 (1%) and A763 (1%). There were no mutation variants identified amongst the efficacy population that were associated with an absence of confirmed responses.
5.2 Pharmacokinetic Properties
Based on Rybrevant monotherapy data, amivantamab exposure increases proportionally over a dose range from 350 to 1750 mg.
Based on the population pharmacokinetics of Rybrevant, steady-state concentrations of Rybrevant were reached by week 13 for both the approved (3-week and 2-week) dosing regimens and the systemic accumulation was 1.9-fold.
Distribution.
The mean (± SD) volume of distribution of amivantamab is 5.34 (± 1.81) L.
Excretion.
The geometric mean (% CV) linear clearance (CL) and terminal half-life associated with linear clearance estimated from population PK parameters were 0.266 L/day (30.4%), and 13.7 days (31.9%), respectively.
Special populations.
Paediatrics (17 years of age and younger).
The pharmacokinetics of Rybrevant in paediatric patients have not been investigated.
Elderly (65 years of age and older).
No clinically meaningful differences in the pharmacokinetics of amivantamab were observed based on age (21-88 years).
Renal impairment.
No clinically meaningful effect on the pharmacokinetics of amivantamab was observed in patients with mild (creatinine clearance [CrCl] of 60 to < 90 mL/min) and moderate (CrCl of 29 to < 60 mL/min) or severe (15 ≤ CrCl < 29 mL/min) renal impairment. Data in patients with severe renal impairment are limited (n=1), but there is no evidence to suggest that dose adjustment is required in these patients. No data are available in patients with end stage renal disease (CrCl < 15 mL/min).
Hepatic impairment.
No clinically meaningful effect on the pharmacokinetics of amivantamab was observed based on mild hepatic impairment [(total bilirubin ≤ ULN and AST > ULN) or (ULN < total bilirubin ≤ 1.5 x ULN and any AST) or moderate hepatic impairment (1.5 x ULN < total bilirubin ≤ 3 x ULN and any AST). Data in patients with moderate hepatic impairment are limited (n=1), but there is no evidence to suggest that dose adjustment is required in these patients. No data are available in patients with severe hepatic impairment (total bilirubin > 3 x ULN and any AST).
Changes in hepatic function are not expected to affect amivantamab elimination since IgG1-based molecules are not metabolised through hepatic pathways.
Sex.
Amivantamab clearance was 24% higher in males than in females, but no clinically meaningful pharmacokinetic differences were observed between male and female patients.
Weight.
The central volume of distribution and clearance of amivantamab increased with increasing body weight. Amivantamab exposure was 30-40% lower in patients who weighed ≥ 80 kg compared to patients with body weight < 80 kg at the same dose. Similar amivantamab exposures were achieved at the recommended dose of Rybrevant in patients with a body weight < 80 kg who received 1050 mg and patients with a body weight ≥ 80 kg who received 1400 mg.
5.3 Preclinical Safety Data
Genotoxicity.
As amivantamab is a monoclonal antibody, genotoxicity studies have not been conducted. Large protein molecules are not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity.
As amivantamab is a monoclonal antibody, carcinogenicity studies have not been conducted.6 Pharmaceutical Particulars
6.1 List of Excipients
Disodium edetate, histidine, histidine hydrochloride monohydrate, methionine, polysorbate 80, sucrose, water for injections.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in Section 4.2 Dose and Method of Administration.
6.3 Shelf Life
Unopened vials.
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
After dilution.
Since amivantamab solutions do not contain a preservative, the product should be used immediately. Administer diluted solutions within 10 hours (including infusion time) at room temperature (15°C to 25°C) and in room light.
6.4 Special Precautions for Storage
Store in a refrigerator at 2°C to 8°C.
Do not freeze.
Store in the original package in order to protect from light.
For storage conditions after dilution of the medicinal product, see Section 6.3 Shelf Life.
6.5 Nature and Contents of Container
Type 1 glass vial with butyl rubber elastomer stopper and aluminium seal with a flip off cap.
Rybrevant is available in cartons containing 1 single-use vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
No data available.
7 Medicine Schedule (Poisons Standard)
S4 Prescription Medicine.
Summary Table of Changes
