What is in this leaflet
This leaflet answers some common questions about Sandrena. It does not contain all the available information. It does not take the place to talking to your doctor or pharmacist.
Keep this information with the pack. You may wish to read it again.
General advice
- This medicine has been prescribed only for your current medical problem. It should not be used for other medical conditions.
- Never give your medicine to anyone else and do not use medicines meant for other people.
- Tell every doctor treating you what medicines you are taking. Always carry a medical information card stating which medicines you are using. This can be very important if for example you are involved in an accident.
- Return unused medicines to your pharmacy for disposal.
- Make sure that other people who live with you or who look after you read this information.
A doctor's prescription is required to obtain this medicine.
What Sandrena is used for
During and after menopause the production of sex hormones produced by the body decreases. Women may then suffer from complaints such as hot flushes, night sweats, vaginal irritation, depression, and loss of sexual desire. Sandrena can be used for the short-term relief of menopausal complaints.
It can also relieve these symptoms in women who have had their ovaries removed. Relief of symptoms usually starts within a few weeks, but optimal results are obtained after three months of treatment. Sandrena is not intended for contraceptive use.
If you have not had a hysterectomy, your doctor will probably combine the Sandrena treatment with another hormone product, a progestogen.
Before you use it
Sandrena may not be suitable for you if you suffer from certain medical conditions.
Before you take it
Tell your doctor if you-
- are pregnant or think you may be pregnant
- have a tumour (e.g. a breast tumour or a tumour in your womb)
- have or ever had heart disease or blood vessel problems
- have or have ever had thrombosis (blood clots)
- recently had unexpected vaginal bleeding
- have or have ever had liver disease
- have uterine fibroids. Careful examinations should be performed, by your doctor, at regular intervals during therapy
- had heart failure, kidney problems or severe hypertension.
Also tell your doctor if:
- you have or have ever had too much cholesterol or other fatty substances in the blood
- you have been treated with other sex hormones recently.
Tell your doctor if you are or think you are allergic to any of the components of the gel. The ingredients are listed at the end of this leaflet. Note this product contains propylene glycol and may cause skin irritation.
Your doctor will conduct a complete gynaecological examination before commencement with Sandrena.
Breast cancer:
Before starting with hormone replacement therapy (HT) you have to inform your doctor of your personal and family medical history. You will get a general and gynaecological examination. You will also get periodic check-ups, especially examinations of the breasts. Every woman is at risk of getting breast cancer, whether or not she takes HT. Breast cancer has been found slightly more often in women using HT than in women of the same age who have never used HT.
It is not known whether Sandrena is associated with the same higher chance of having breast cancer diagnosed as other hormone replacement therapies.
Nevertheless, if you are concerned about the risk of breast cancer, discuss the risk compared to the benefits of treatment with your doctor.
Stroke:
There may be an increased risk of stroke when using this product.
Dementia:
There may be an increased risk of dementia when using this product.
Ovarian/Endometrial cancer:
Ovarian cancer (cancer of the ovaries) is very rare, but it is a serious condition. It can be difficult to diagnose, because there are often no obvious signs of the disease.
Evidence from studies suggests an increased risk in women taking oestrogen-only or combined oestrogen-progestogen HRT, which becomes apparent within 5 years of use and diminishes over time after stopping.
Some other studies, including the Women's Health Initiative (WHI) trial, suggest that use of combined HRTs may be associated with a similar risk.
Women who still have a uterus must take both estrogen and progestogen as part of HT. This is because estrogen stimulates the growth of the lining of the uterus (called the endometrium). Before menopause this lining is shed during your period through the action of a natural progestogen.
After menopause, taking estrogen on its own as HT may lead to irregular bleeding and a disorder of the uterus lining called endometrial hyperplasia, which can become endometrial cancer. Progestogens help protect the lining of the uterus from developing this disorder.
Also, clinical studies on similar products showed a possible increased risk of cardiovascular problems in the first year of use and no benefit thereafter.
Thrombosis (blood clots):
All women have a very small chance of having a blood clot in the veins of the leg, lung or other parts of the body whether or not they take HT. Using some forms of HT may slightly increase this small chance. Whether or not Sandrena may increase the chance of having a blood clot is not known.
You are more likely to have a blood clot (whether or not you use HT) if:
- you are very overweight;
- you have had a blood clot in the veins of your legs or in your lungs before;
- blood clots run in your family;
- you have systemic lupus erythematosus (a disease of your immune system);
- you are unable to move for long periods, for example after a long illness or major operation;
- may be also if you have varicose veins.
You should talk to your doctor about whether you should use Sandrena if any of these apply to you.
If you get a blood clot while you are using Sandrena you should stop taking it immediately and contact your doctor.
Warning signs to look out for are:
- unusual pains or swelling of your legs;
- pain in your chest or sudden shortness of breath.
Other medicines may influence the effects of Sandrena, or Sandrena may affect other medicines.
You must tell your doctor or pharmacist if you are taking (or intend to take) other medicines such as:
- anticonvulsants, barbiturates, carbamazepine and hydantoins (medicines for epilepsy or sleeplessness);
- rifampicin, rifabutin, nevirapine, efavirenz (medicines for infections);
- meprobamate and phenylbutazone
- ritonavir, nelfinavir;
- herbal preparations containing St John’s Wort.
These drugs may reduce the effects of Sandrena. Sandrena may also reduce the effectiveness of other medicines such as some oral hypoglycaemics (medicines which lower blood sugar), antihypertensives (medicines which lower blood pressure) and some anticoagulants (medicines which "thin" the blood). Let your pharmacist or doctor know if you are a diabetic, or are being treated for blood clotting or high blood pressure.
This is not a complete list of medicines which may interact with Sandrena.
Please talk to your doctor or pharmacist.
Do not use Sandrena if you are pregnant or breast-feeding, or think you may be pregnant.
Yearly medical checks are recommended. Additionally, the doctor may check with you on how the treatment is progressing every six months or so.
How to use it
Apply Sandrena as directed by your doctor. You should also read the instructions on the label of your medicine.
If you are not sure how to use Sandrena ask your doctor or pharmacist.
How much to use
The usual starting dose is one sachet per day. Your doctor or pharmacist will tell you exactly how much to use.
How to use it
Open the sachet(s). Rub the contents daily on the skin of your lower abdomen or thighs. The application surface area should be one to two times the size of the hand.
Sandrena should not be applied on the breasts, face or irritated skin.
Avoid contact with eyes. If Sandrena does get into your eyes, wash out thoroughly with water. Wash hands thoroughly immediately after application.
In clinical trials the use of Sandrena has infrequently caused irritation of the skin. The chance of skin reactions can be further decreased by changing the area of application daily (e.g. left and right side on alternate days).
Sandrena is used either cyclically or continuously, in individually adjusted doses.
If you have not had a hysterectomy, your doctor will probably prescribe a progestogen, to help prevent overgrowth of the tissues lining your womb.
What to do if you miss a dose
If you forget to apply the gel do so as soon as you remember, unless you are more than twelve hours late. If you are more than twelve hours late, do not apply the missed dose and just carry on with the next dose as normal.
While you are using it
Ability to drive or operate machinery. As far as is known, Sandrena has no effect on alertness and concentration.
Discuss with your doctor or pharmacist if you are unsure of any of the aspects of this product.
Side effects
Side effects, which may or may not be related to Sandrena are most common in the first months of the treatment. They are usually mild leading only seldom to discontinuation of the treatment.
Common (more than 1/100):
- breast pain or tension;
- headache;
- fatigue;
- nausea;
- vomiting;
- stomach cramps;
- flatulence;
- dizziness;
- palpitations;
- swelling from water retention;
- weight increase;
- varicose veins;
- vaginal discharge;
- lethargy;
- depression;
- nervousness;
- hot flushes;
- itch of application site;
- pain;
- increased sweating;
- unscheduled or breakthrough bleedings.
Uncommon (less than 1/100):
- skeletal pain;
- itching;
- muscle pain & cramps;
- nervousness;
- changes in mood or libido;
- migraine;
- benign breast or endometrial tumours;
- increased appetite;
- excessive growth of the cells of the uterus.
Tell your doctor if spotting occurs or if any side-effects become troublesome or continue.
It is also important to tell your doctor or pharmacist if you experience any other unusual or unexpected symptoms during treatment with Sandrena.
Seek advice before using other medications to deal with any side effects you may have.
You should stop using Sandrena and seek medical attention immediately if you experience any signs of:
- thrombosis (red, painful or swollen leg, difficulty breathing, chest pain, headache or pain elsewhere in your body, dizziness, fainting, disturbances in vision, swollen ankles);
- jaundice (yellowing of the eyes or skin).
Other side effects not listed above may also occur in some people.
Overdose
If someone has taken or used several sachets at once, there is no need for great concern. However, you should consult a doctor.
Symptoms that may arise are nausea and vomiting. Vaginal bleeding may occur after a few days.
After taking it
Storage
Keep your Sandrena gel in the original container in a safe place out of the reach of children.
Store below 25°C.
The expiry date (sometimes written as "exp") is also printed on the strip of sachets - do not use after this date.
Disposal
Return any unused medicine to your pharmacist
Product description
What it looks like
Sandrena is a smooth opalescent gel.
It is available in 0.5 g or 1 g sachets in packs containing28 or 91 sachets.
* Not all presentation and/or pack sizes are available.
Ingredients
Sandrena consists of 0.1% estradiol.
Each 1 g sachet of gel contains 1.0 mg of estradiol.
Each 0.5 g sachet of gel contains 0.5 mg of estradiol.
It also contains the following inactive ingredients:
- carbomer 934P;
- trolamine;
- propylene glycol;
- ethanol;
- purified water.
Sponsor
Orion Pharma (Aus) Pty Limited
Level 24, Tower 3
300 Barangaroo Avenue, Sydney,
NSW 2000, Australia
Telephone: 1800 861 913
AUST R: 93609 (1.0 g sachet)
AUST R: 93608 (0.5g sachet)
This leaflet was revised in July 2022.
Published by MIMS September 2022
 In clinical trials dermal irritation has occurred in less than 4% of the patients.
In clinical trials dermal irritation has occurred in less than 4% of the patients.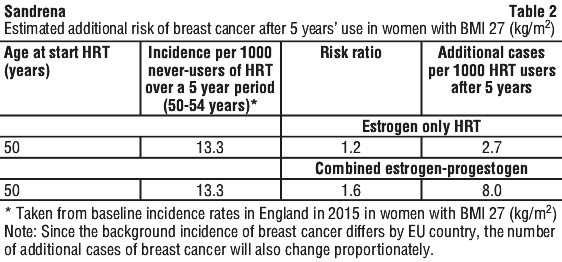




 During Sandrena treatment the estradiol/estrone ratio remains at 0.7, while during peroral estrogen treatment it usually drops to less than 0.2. The steady-state bioavailability of Sandrena is 82 per cent compared to the equivalent oral dose of estradiol valerate.
During Sandrena treatment the estradiol/estrone ratio remains at 0.7, while during peroral estrogen treatment it usually drops to less than 0.2. The steady-state bioavailability of Sandrena is 82 per cent compared to the equivalent oral dose of estradiol valerate. Chemical name: 17β-estradiol hemihydrate; estra-1, 3, 5 (10)-triene-3, 17β-diol. 0.5 H2O.
Chemical name: 17β-estradiol hemihydrate; estra-1, 3, 5 (10)-triene-3, 17β-diol. 0.5 H2O.