


What is in this leaflet
This leaflet answers some common questions about Simvastatin Sandoz. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risk of you taking this medicine against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Simvastatin Sandoz is used for
Simvastatin Sandoz helps to lower cholesterol and triglyceride levels.
Simvastatin Sandoz is used in people who have Coronary Heart Disease (CHD) or who are at high risk of CHD, for example if they have diabetes, a history of stroke or other blood vessel disease.
Simvastatin Sandoz may be used in these people, regardless of their cholesterol level, to
- help prolong life by reducing the risk of a heart attack
- reduce the risk of stroke
- reduce the need for surgery to increase blood flow to the heart
- reduce the need for hospitalisation due to angina.
Cholesterol
Everyone has cholesterol and triglycerides in their blood. They are types of blood fat needed by the body for many things, such as building cell walls, making bile acids (which help to digest food) and certain hormones. However, too much cholesterol can be a problem.
Your body makes cholesterol, but it also comes from food.
Normally the body balances the cholesterol it makes with the cholesterol it gets from food. This means if more cholesterol comes from food, less is made by the body. However, if you eat a diet high in fat, your body may not keep this balance and your cholesterol levels rise.
High cholesterol is more likely to occur with certain diseases or if you have a family history of high cholesterol.
When you have high levels of cholesterol it may 'stick' to the inside of your blood vessels instead of being carried to the parts of the body where it is needed. Over time, this can form hard areas, called plaque on the walls of blood vessels, making it more difficult for the blood to flow. This blocking of your blood vessels can lead to Coronary Heart Disease (such as heart attack and angina), and stroke.
In patients with CHD, Simvastatin Sandoz may slow down the hardening of blood vessels and reduces the risk of developing new plaques.
There are different types of cholesterol, called LDL and HDL. LDL cholesterol is the 'bad' cholesterol that can block your blood vessels. HDL cholesterol, on the other hand, is the 'good' cholesterol that is thought to remove the 'bad' cholesterol from the blood vessels.
Triglycerides
Triglycerides are a source of energy for the body. However, as with cholesterol, too much triglycerides can be a problem.
How Simvastatin Sandoz works
Simvastatin Sandoz belongs to a group of medicines called HMG-CoA reductase inhibitors. It works by reducing the amount of cholesterol made by the liver. In terms of good and bad cholesterol, Simvastatin Sandoz reduces the 'bad' cholesterol and raises the 'good' cholesterol.
Simvastatin Sandoz does not reduce the cholesterol and triglycerides that come from fat in food.
Therefore, when you are taking Simvastatin Sandoz, you also need to follow a low fat diet and other measures, such as exercise and weight control.
In most people, there are no symptoms of high cholesterol or triglycerides. Your doctor can measure your cholesterol and triglycerides with a simple blood test.
Safety and effectiveness have been studied in 10-17 year old boys and in girls, who had started their menstrual period at least one year before (see How to take Simvastatin Sandoz). Simvastatin Sandoz has not been studied in children under the age of 10 years. For more information, talk to your doctor.
Ask your doctor if you have any questions about why this medicine was prescribed for you. Your doctor may have prescribed it for another reason.
Simvastatin Sandoz is not addictive.
Before you take Simvastatin Sandoz
When you must not take it
Do not take this medicine if:
- you have an allergy to Simvastatin Sandoz or other brands of simvastatin or to any of the active ingredient listed at the end of this leaflet.
Symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body, skin rash, itchiness or painful joints.
Do not take this medicine if you have or have had any of the following medical conditions:
- liver disease
- muscle pain, tenderness or weakness from other medicines used to treat high cholesterol or triglycerides.
Do not take this medicine if you are pregnant or breastfeeding. Your baby may absorb this medicine in the womb or from breast milk and therefore there is a possibility of harm to the baby.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal. If you take this medicine after the expiry date has passed, it may not work.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if:
- you intend to become pregnant or plan to breast feed. Simvastatin Sandoz should not be used during pregnancy or while breast-feeding.
- you have unexplained muscle pain, tenderness or weakness not caused by exercise. This is because on rare occasions, muscle problems can be serious, including muscle breakdown resulting in kidney damage that can lead to death. Your doctor may do a blood test to check for certain muscle problems.
- you are Asian.
- you ever had liver disease. Your doctor will do a blood test to make sure you have no problems with your liver.
- you have kidney disease or any other medical problems
- you drink alcohol regularly
- you have allergies to any other medicines, or any other substances, such as foods, preservatives or dyes.
- you have or have had myasthenia gravis (a disease causing general muscle weakness including in some cases muscles used for breathing) or ocular myasthenia (a disease causing eye muscle weakness) as statins may lead to occurrence of myasthenia or aggravate the condition.
Simvastatin Sandoz contains lactose. If you have been told by your doctor that you have intolerance to some sugars, tell your doctor before taking it.
If you have not told your doctor about any of the above, tell them before you start taking Simvastatin Sandoz.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines should not be taken with Simvastatin Sandoz as they may increase the risk of muscle side effects with Simvastatin Sandoz. It is particularly important to tell your doctor if you are taking:
- nefazodone, used to treat depression
- medicines containing cobicistat (a drug used in the treatment of HIV infection)
- protease inhibitors, used to treat HIV infection, including indinavir, nelfinavir, ritonavir and saquinavir
- certain hepatitis C virus protease inhibitors (such as boceprevir or telaprevir)
- gemfibrozil, used to treat high cholesterol levels
- ciclosporin, used to suppress the immune system
- danazol
- erythromycin, clarithromycin, telithromycin and fusidic acid, antibiotics used to treat infections
- ketoconazole, itraconazole, fluconazole, posaconazole and voriconazole, used to treat certain fungal infections.
If you are taking any of the medicines listed above, your doctor may suggest stopping Simvastatin Sandoz temporarily or permanently.
Some medicines and Simvastatin Sandoz interfere with each other. Because taking Simvastatin Sandoz with any of the following drugs can increase the risk of muscle problems (see Side Effects), it is particularly important to tell your doctor if you are taking:
- other medicines used to lower cholesterol levels, e.g. other fibrates (such as gemfibrozil) or nicotinic acid (also known as niacin)
- warfarin, ticagrelor or other drugs used to prevent blood clots
- colchicine, used to treat gout
- verapamil, diltiazem or amlodipine, used to treat high blood pressure, angina or other heart conditions
- lomitapide (a drug used to treat a serious and rare genetic cholesterol condition)
- amiodarone, used to treat irregular heartbeat.
- digoxin, used to treat heart failure
- certain hepatitis C antiviral agents, such as elbasvir or grazoprevir
- daptomycin, a drug used to treat complicated skin and skin structure infections and bacteraemia.
These medicines may be affected by Simvastatin Sandoz, or may affect how well it works or may increase the risk of side effects with Simvastatin Sandoz. You may need to take different amounts of your medicine, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
You should also tell any doctor who is prescribing a new medication for you that you are taking Simvastatin Sandoz.
How to take Simvastatin Sandoz
How much to take
Take Simvastatin Sandoz only when prescribed by your doctor. Your doctor will tell you how many tablets you need to take each day. This depends on your cholesterol and triglyceride levels and other factors, such as kidney disease.
For adults, the recommended starting dose is 10 mg or 20 mg per day, taken in the evening. To have the best effect, this may need to be increased up to 80 mg daily to have the best effect.
Because of the increased risk of muscle problems, the 80 mg dose is only for patients at high risk of heart disease problems who have not reached their cholesterol goal on the lower doses.
For people with CHD or risk factors for CHD, the usual starting dose is 40 mg per day, taken in the evening.
For children (10-17 years old), the recommended usual starting dose is 10 mg a day in the evening. The maximum recommended dose is 40 mg a day.
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
Ask your doctor or pharmacist if you are unsure of the correct dose for you. They will tell you exactly how much to take.
How to take it
Swallow the tablets with a full glass of water.
If you need to break Simvastatin Sandoz, hold the tablet with both hands and snap along the break line.
When to take Simvastatin Sandoz
Take your medicine once a day in the evening. The liver produces its greatest amount of cholesterol when the body is at rest and when there is no dietary intake. For most people this is at night when they are asleep. Therefore, Simvastatin Sandoz is more effective when taken in the evening. A good time would be after your evening meal. However, it does not matter whether you take it before or after food.
Take your medicine at about the same time each day. Taking it at the same time each evening will have the best effect. It will also help you remember when to take the tablets.
How long to take Simvastatin Sandoz
Your doctor will determine how long you have to take Simvastatin Sandoz.
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to lower your cholesterol levels, but does not cure your condition. Therefore, you must continue to take it as directed by your doctor if you expect to lower your cholesterol and keep it down. You may have to take cholesterol-lowering medicines for the rest of your life. If you stop taking this medicine, your cholesterol levels may rise again.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to. Otherwise, take it as soon as you remember, and then go back to taking your tablet as would normally.
Do not take a double dose to make up for the dose you missed. This may increase the chance of getting an unwanted side effect.
If you are not sure whether to skip the dose, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at your nearest hospital, if you think you or anyone else has taken too much Simvastatin Sandoz. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking Simvastatin Sandoz
Things you must do
If you become pregnant while taking Simvastatin Sandoz, stop taking it and contact your doctor immediately.
Have your blood fats checked when your doctor says so, to make sure this medicine is working.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Simvastatin Sandoz.
If you are about to have elective surgery, tell your doctor that you are taking Simvastatin Sandoz. Your doctor may suggest stopping the tablets a few days before surgery.
Things you must not do
Do not stop taking Simvastatin Sandoz without your doctor's permission.
Do not take Simvastatin Sandoz to treat any other complaint unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Things to be careful of
Be careful driving or operating machinery until you know how Simvastatin Sandoz affects you. Simvastatin Sandoz generally does not cause any problems with your ability to drive a car or operate machinery. However, as with many other medicines, Simvastatin Sandoz may cause dizziness in some people. Make sure you know how you react to Simvastatin Sandoz before you drive a car or operate machinery.
Avoid drinking large quantities of alcohol. Drinking large quantities of alcohol may increase your chance of Simvastatin Sandoz causing liver problems.
Grapefruit juice should be avoided while taking Simvastatin Sandoz. Grapefruit juice contains one or more components that alter the metabolism of some medicines, including Simvastatin Sandoz.
Changes to lifestyle that may help reduce the chance of coronary heart disease
Lowering high cholesterol can help reduce your chances of having Coronary Heart Disease (CHD). However, your chances of having CHD may be increased by several other factors including high blood pressure, cigarette smoking, diabetes, excess weight, a family history of CHD, being a male and being a woman who has reached menopause.
Some self help measures suggested below may help your condition and help reduce your chances of having CHD. Talk to your doctor, pharmacist, or dietician about these measures and for more information.
Diet - continue the low fat diet recommended by your doctor, dietician or pharmacist.
Weight - your doctor may advise you to lose weight if you are overweight.
Exercise - make exercise a part of your routine - walking is good. Ask your doctor for advice before starting exercise.
Smoking - your doctor will advise you to stop smoking or at least cut down.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Simvastatin Sandoz.
Simvastatin Sandoz helps most people with high cholesterol, but it may have unwanted side effects in some people. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- constipation, diarrhoea, wind
- stomach upset or pain, feeling sick (nausea)
- headache
- dizziness
These are the more common side effects of Simvastatin Sandoz. For the most part, these are mild and short-lived.
Tell your doctor immediately if you notice any of the following:
- aching muscles, muscle tenderness or weakness, not caused by exercise (in very rare cases this may not go away after stopping Simvastatin Sandoz)
- brown/black coloured urine.
On rare occasions, muscle problems can be serious, including muscle breakdown resulting in kidney damage that can lead to death.
The risk of muscle problems is greater for:
- patients taking higher doses of Simvastatin Sandoz, particularly the 80 mg dose
- older patients (65 years of age and older)
- female patients
- patients with abnormal kidney function
- patients with thyroid problems.
Tell your doctor immediately if you notice any of the following:
- tingling in the hands or feet
- signs of anaemia, such as tiredness, being short of breath, and looking pale
- larger breasts than normal in men
- bruising more easily than normal
- skin rash, itchiness
- fever, generally feeling unwell
- pinkish, itchy swellings on the skin, also called hives or nettle rash
- painful, swollen joints
- weakness in your arms or legs that worsens after periods of activity, double vision or drooping of your eyelids, difficulty swallowing, or shortness of breath (symptoms of myasthenia).
These may be serious side effects of Simvastatin Sandoz. You may need urgent medical attention. Serious side effects are rare.
If any of the following happen, stop taking Simvastatin Sandoz and tell your doctor immediately, or go to Accident and Emergency at your nearest hospital:
- swelling of the limbs, face, lips, mouth or throat which may cause difficulty in swallowing or breathing
- shortness of breath
- itchy rash or hives.
These are serious side effects. You may need urgent medical attention or hospitalisation. Serious side effects are rare.
Also, tell your doctor if you notice:
- hair loss
- muscle cramps
- trouble sleeping
- poor memory, memory loss, confusion
- feelings of depression
- blurred vision and impaired vision
- erectile dysfunction
- breathing problems including persistent cough and/or shortness of breath or fever.
- gynaecomastia (breast enlargement in men) (very rare)
- muscle rupture (very rare)
- rash that occur on the skin or sores in the mouth (lichenoid drug eruptions) (very rare)
These are other side effects that have been reported with Simvastatin Sandoz.
Liver problems can also occur and may be serious. Your doctor will do regular blood tests to check your liver.
Tell your doctor immediately if you have the following symptoms of liver problems:
- feel tired or weak
- loss of appetite
- upper belly pain
- dark urine
- yellowing of your skin or the whites of your eyes.
Tell your doctor or pharmacist if you notice any other effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Other side effects not listed above may also occur in some people.
After taking Simvastatin Sandoz
Storage
Keep your medicine in the original container.
If you take it out of its container it may not keep well.
Keep your medicine in a cool dry place where the temperature stays below 25°C. Protect from light.
Do not store Simvastatin Sandoz or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Simvastatin Sandoz 5 mg: yellow, oval, scored, convex, film-coated tablets, coded 'SIM 5' on one side.
Simvastatin Sandoz 10 mg - pale pink, oval, scored, convex, film-coated tablets, coded 'SIM 10' on one side.
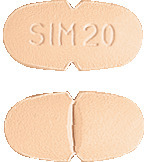
Simvastatin Sandoz 20 mg - orange, oval, scored, convex, film-coated tablets, coded 'SIM 20' on one side.
Simvastatin Sandoz 40 mg - pink, oval, scored, convex, film-coated tablets, coded 'SIM 40' on one side.
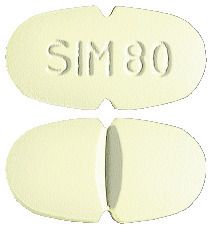
Simvastatin Sandoz 80 mg - light green, oval, scored, convex, film-coated tablets, coded 'SIM 80' on one side.
They are available in blisters of 30 tablets.
Ingredients
Active ingredients:
Simvastatin Sandoz 5 mg - 5 mg simvastatin
Simvastatin Sandoz 10 mg - 10 mg simvastatin
Simvastatin Sandoz 20 mg - 20 mg simvastatin
Simvastatin Sandoz 40 mg - 40 mg simvastatin
Simvastatin Sandoz 80 mg - 80 mg simvastatin
Inactive ingredients:
- pregelatinised maize starch
- lactose monohydrate
- microcrystalline cellulose
- butylated hydroxyanisole
- ascorbic acid
- citric acid monohydrate
- magnesium stearate
- hypromellose
- purified talc
- titanium dioxide
- iron oxide yellow (Simvastatin Sandoz 5 mg, 10 mg and 20 mg only)
- iron oxide red (Simvastatin Sandoz 10 mg, 20 mg and 40 mg only)
- indigo carmine aluminium lake (Simvastatin Sandoz 80 mg only)
- quinoline yellow aluminium lake (Simvastatin Sandoz 80 mg only).
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Supplier
Sandoz Pty Ltd
100 Pacific Highway
North Sydney, NSW 2060
Australia
Tel 1800 726 369
This leaflet was revised in November 2023.
Australian Register Numbers
Simvastatin Sandoz 5 mg film-coated tablets: AUST R 114796 (blisters)
Simvastatin Sandoz 10 mg film-coated tablets: AUST R 114797 (blisters)
Simvastatin Sandoz 20 mg film-coated tablets: AUST R 114798 (blisters)
Simvastatin Sandoz 40 mg film-coated tablets: AUST R 114800 (blisters)
Simvastatin Sandoz 80 mg film-coated tablets: AUST R 98624 (blisters)
® Registered Trade Mark. The trade marks mentioned in this material are the property of their respective owners.
Published by MIMS January 2024

 Myopathy has been reported rarely.
Myopathy has been reported rarely. In a separate study involving 180 patients with combined hyperlipidaemia, simvastatin tablets 10 mg daily for 17 weeks was also shown to be effective in lowering total-C, LDL-C, VLDL-C, TGs and Apo B (see Table 4).
In a separate study involving 180 patients with combined hyperlipidaemia, simvastatin tablets 10 mg daily for 17 weeks was also shown to be effective in lowering total-C, LDL-C, VLDL-C, TGs and Apo B (see Table 4). The data from these studies demonstrate that in patients with hypercholesterolaemia and normal or slightly raised TGs, simvastatin consistently reduce total-C, LDL-C, TGs, VLDL-C and Apo B in a dose dependent manner.
The data from these studies demonstrate that in patients with hypercholesterolaemia and normal or slightly raised TGs, simvastatin consistently reduce total-C, LDL-C, TGs, VLDL-C and Apo B in a dose dependent manner. In the upper dose comparative study, one-third of patients obtained a reduction in LDL-C of 53% or more at the 80 mg dose. The percent reduction in LDL-C was essentially independent of the baseline level. In contrast, the percent reduction in TG was related to the baseline level of TG. Of the 664 patients randomised to 80 mg, 475 patients with plasma TG less than or equal to 2.25 mmol/L had a median reduction in TG of 21%, while in 189 patients with hypertriglyceridaemia (> 2.25 mmol/L), the median reduction in TG was 36%. In these studies, patients with TG > 4.0 mmol/L were excluded.
In the upper dose comparative study, one-third of patients obtained a reduction in LDL-C of 53% or more at the 80 mg dose. The percent reduction in LDL-C was essentially independent of the baseline level. In contrast, the percent reduction in TG was related to the baseline level of TG. Of the 664 patients randomised to 80 mg, 475 patients with plasma TG less than or equal to 2.25 mmol/L had a median reduction in TG of 21%, while in 189 patients with hypertriglyceridaemia (> 2.25 mmol/L), the median reduction in TG was 36%. In these studies, patients with TG > 4.0 mmol/L were excluded. The effects of simvastatin on major vascular events and major coronary events were similar in all subgroups of patients (see Figure 1).
The effects of simvastatin on major vascular events and major coronary events were similar in all subgroups of patients (see Figure 1). The risk reductions produced by simvastatin in both major coronary events and major vascular events were evident and consistent across all baseline characteristics shown in Figure 1. In addition, these risk reductions were evident and consistent regardless of prior treated hypertension, creatinine levels up to the entry limit of 2.3 mg/dL, apolipoprotein A-I and B levels, baseline concomitant cardiovascular medications (i.e. aspirin, beta-blockers, angiotensin converting enzyme (ACE) inhibitors or calcium channel blockers), smoking status, alcohol intake or obesity.
The risk reductions produced by simvastatin in both major coronary events and major vascular events were evident and consistent across all baseline characteristics shown in Figure 1. In addition, these risk reductions were evident and consistent regardless of prior treated hypertension, creatinine levels up to the entry limit of 2.3 mg/dL, apolipoprotein A-I and B levels, baseline concomitant cardiovascular medications (i.e. aspirin, beta-blockers, angiotensin converting enzyme (ACE) inhibitors or calcium channel blockers), smoking status, alcohol intake or obesity.

 Simvastatin compared with placebo showed -31.8% vs +1.3% change in LDL level after 8 weeks on 10 mg and -36.4% vs -2.9% after 8 weeks on 20 mg.
Simvastatin compared with placebo showed -31.8% vs +1.3% change in LDL level after 8 weeks on 10 mg and -36.4% vs -2.9% after 8 weeks on 20 mg. A single dose of 2 g niacin extended release coadministered with 20 mg simvastatin increased the AUC and Cmax of simvastatin acid by approximately 60% and 84%, respectively, compared to administration of 20 mg simvastatin alone. In this study, the effect of simvastatin on niacin pharmacokinetics was not measured.
A single dose of 2 g niacin extended release coadministered with 20 mg simvastatin increased the AUC and Cmax of simvastatin acid by approximately 60% and 84%, respectively, compared to administration of 20 mg simvastatin alone. In this study, the effect of simvastatin on niacin pharmacokinetics was not measured. Chemical name: [1S-[1α, 3α, 7β, 8β (2S, 4S*),8aβ]]-1,2,3,7,8,8a-hexahydro-3,7-dimethyl-8-[2-(tetrahydro-4-hydroxy-6-oxo-2H-pyran-2-yl)ethyl]-1-naphthalenyl 2,2-dimethylbutanoate.
Chemical name: [1S-[1α, 3α, 7β, 8β (2S, 4S*),8aβ]]-1,2,3,7,8,8a-hexahydro-3,7-dimethyl-8-[2-(tetrahydro-4-hydroxy-6-oxo-2H-pyran-2-yl)ethyl]-1-naphthalenyl 2,2-dimethylbutanoate.