What is in this leaflet
This leaflet answers some common questions about SKYRIZI. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using SKYRIZI against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Read this leaflet carefully before you start using this medicine. It includes important information on the safe and effective use of this medicine.
Keep this leaflet with the medicine. You may need to read it again.
What SKYRIZI is used for
SKYRIZI is used to treat adults with moderate to severe plaque psoriasis, an inflammatory condition affecting the skin and nails.
SKYRIZI contains the active ingredient risankizumab, which is a type of protein called a monoclonal antibody. It works by stopping a protein in the body called IL-23 which causes inflammation.
SKYRIZI reduces the inflammation which improves skin clearance and the appearance of the nails. It also reduces symptoms of psoriasis such as burning, itching, pain, redness and scaling.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not expected to affect your ability to drive a car or operate machinery.
Before you use SKYRIZI
When you must not use it
Do not use this medicine if you have an allergy to risankizumab or any of the other ingredients listed at the end of this leaflet.
Do not use this medicine if you have an active infection which your doctor thinks is important.
Do not use this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start using this medicine, talk to your doctor.
Before you start to use it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor, pharmacist or nurse before and during use of SKYRIZI if you have or have had any of the following medical conditions:
- you currently have an infection or if you have an infection that keeps coming back.
- you have tuberculosis (TB).
- you have recently received or plan to receive an immunisation (vaccine). You should not be given certain types of vaccines while using SKYRIZI.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you are pregnant, think you may be pregnant or are planning to have a baby, ask your doctor for advice before using this medicine. This is because it is not known how this medicine will affect the baby.
If you are breast-feeding or are planning to breast-feed, talk to your doctor before using this medicine.
If you have not told your doctor about any of the above, tell him/her before you start using SKYRIZI.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
How to use SKYRIZI
Follow all directions given to you by your doctor, pharmacist or nurse carefully.
If you do not understand the instructions on the box, ask your doctor, pharmacist or nurse for help.
How much to use
SKYRIZI is given as 2 injections under your skin (called 'subcutaneous injections').
The dose is 150 mg given as two 75 mg injections.
- 1st dose = 150 mg (two 75 mg injections) as directed by your doctor, pharmacist or nurse
- 2nd dose = 150mg (two 75 mg injections) 4 weeks after the 1st dose
- Further doses = 150mg (two 75 mg injections) every 12 weeks starting after the 2nd dose
How to use it
You and your doctor, pharmacist or nurse will decide if you should inject SKYRIZI yourself. Do not inject yourself with this medicine unless you have been trained by your doctor, pharmacist or nurse. A caregiver may also give your injections after training.
Special disposal container can be obtained from your local pharmacy.
Read the 'Instructions for Use' before injecting SKYRIZI yourself.
Instructions for Use
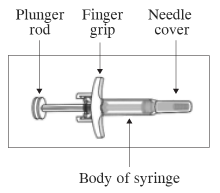
Important information to know before you inject SKYRIZI.
- SKYRIZI is for single use in one patient only. Discard any residue.
- Receive training on how to inject SKYRIZI before giving an injection. Talk to your doctor, pharmacist or nurse if you need help.
- Mark the dates on your calendar so you know when to next use SKYRIZI.
- Keep SKYRIZI in the original carton to protect from light until it is time to use it.
- Do not inject if the liquid is cloudy or contains flakes or large particles. The liquid should look clear to slightly yellow and may contain tiny white or clear particles.
- Do not use if the expiration date has passed.
- Do not use if the liquid has been frozen (even if thawed).
- Do not shake the syringe.
- Do not use if the syringe has been dropped or damaged.
- Do not use if the syringe tray cover is broken or missing. Return this medicine to the pharmacy.
- Do not remove the needle cover until just before the injection.
For a more comfortable injection:
Take the carton out of the refrigerator and leave it at room temperature, out of direct sunlight, for 15 to 30 minutes before injecting. - Do not remove the syringes from the carton until ready to inject.
- Do not warm SKYRIZI in any other way. For example, do not warm it in a microwave or in hot water.
Follow the steps below each time you use SKYRIZI.
STEP 1
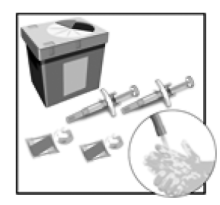
Place the following on a clean, flat surface:
- 2 pre-filled syringes and 2 alcohol pads (provided).
- 2 cotton balls or gauze pads (not provided).
- Special disposal container (not provided).
Wash and dry your hands.
Start with one syringe for the first injection.
For a full dose, 2 injections are required, one after the other.
STEP 2
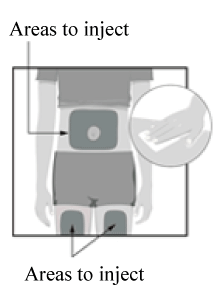
Choose from these 3 areas to inject:
- Front of left thigh.
- Front of right thigh.
- Belly (abdomen) at least 5 cm from the belly button (navel).
For the second syringe, inject at least 3 cm away from the first injection.
- Do not inject into the same place.
Before each injection, wipe where you will inject in a circular motion with an alcohol pad. - Do not touch or blow on the injection site after it is cleaned. Allow the skin to dry before injecting.
- Do not inject through clothes.
- Do not inject into skin that is sore, bruised, red, hard, scarred or has stretch marks.
- Do not inject into areas affected by psoriasis.
STEP 3
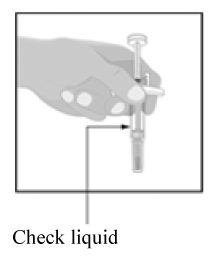
Hold the syringe with covered needle pointing down, as shown.
Check the liquid in the syringe.
- It is normal to see bubbles in the window.
- The liquid should look clear to slightly yellow and may contain tiny white or clear particles.
- Do not use if the liquid is cloudy or contains flakes or large particles.
STEP 4
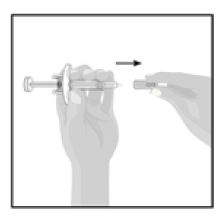
Removing the needle cover:
- Hold the syringe in one hand between the finger grip and needle cover.
- With the other hand, gently pull the needle cover straight off.
- Do not hold or pull plunger rod when removing the needle cover.
- You may see a drop of liquid at the end of the needle. This is normal.
- Throw away the needle cover.
- Do not touch the needle with your fingers or let the needle touch anything.
STEP 5
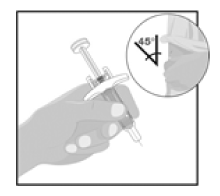
Hold the body of the syringe in one hand between the thumb and index finger, like you would a pencil.
Gently pinch the area of cleaned skin with your other hand and hold it firmly.
Insert the needle all the way into the skin at about a 45-degree angle using a quick, short movement. Keep the syringe steady at the same angle.
STEP 6
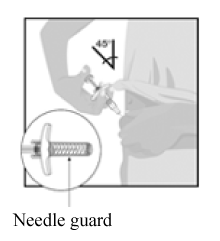
Slowly push the plunger rod all the way in until all of the liquid is injected.
Pull the needle out of the skin while keeping the syringe at the same angle.
Stop pinching the skin around your injection site.
Slowly take your thumb off the plunger rod. The needle will then be covered by the needle guard.
- The needle guard will not activate unless all the liquid is injected.
- Speak to your doctor, pharmacist or nurse if you think you may have not given a full dose.
Press a cotton ball or gauze pad where you have injected and hold for 10 seconds.
Do not rub the skin where you have injected. You may have slight bleeding from where you injected. This is normal.
STEP 7
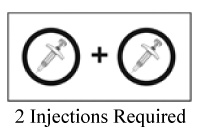
For a full dose, two injections are needed, one after the other.
- Repeat Steps 2 through 6 with the second syringe.
- Inject the second syringe straight after the first injection but at least 3 cm away from the first injection.
STEP 8
Throw away used syringes in a special disposal container right away after use.
- Do not throw away used syringes in the household waste.
- Your doctor, pharmacist or nurse will tell you how to return the full special disposal container.
How long to use it
Do not stop using SKYRIZI without talking to your doctor first. If you stop treatment, your symptoms may come back.
If you forget to use it
If you forget to use SKYRIZI, inject a dose as soon as you remember. Talk to your doctor or pharmacist if you are not sure what to do.
If you use too much (overdose)
If you have used more SKYRIZI than you should, or the dose has been given sooner than prescribed, talk to your doctor.
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have used too much SKYRIZI. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using SKYRIZI
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are using SKYRIZI.
Tell any other doctors, dentists, and pharmacists who treat you that you are using this medicine.
If you become pregnant while using this medicine, tell your doctor immediately.
Things you must not do
You should not receive a live vaccine while taking SKYRIZI. Talk to your doctor, pharmacist or nurse before receiving any vaccination while taking SKYRIZI. Do not use SKYRIZI to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop using your medicine or lower the dosage without checking with your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are using SKYRIZI.
This medicine helps most people, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Talk to your doctor or get medical help as soon as possible if you have symptoms of serious infection such as:
- Fever, flu-like symptoms, night sweats.
- Feeling tired or short of breath, cough which will not go away.
- Warm, red and painful skin, or a painful skin rash with blisters.
Your doctor will decide if you can keep using SKYRIZI.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- Upper respiratory infections with symptoms such as a sore throat and stuffy nose
- Feeling tired
- Fungal skin infection
- Injection site reactions
- Headache
- Small raised red bumps on the skin.
The above list includes the more common side effects of SKYRIZI.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using SKYRIZI
Storage
Keep this medicine out of the sight and reach of children.
Do not use this medicine after the expiration date which is stated on the syringe label and outer carton after 'EXP'.
Store SKYRIZI at 2°C to 8°C. Refrigerate. Do not freeze.
Keep the pre-filled syringes in the original carton in order to protect from light.
The liquid should look clear to slightly yellow. The liquid may contain tiny white or clear particles. Do not use if the liquid is cloudy or contains flakes or large particles.
Disposal
After injecting SKYRIZI, immediately throw away the used syringes in a special container as instructed by your doctor, nurse or pharmacist.
If your doctor tells you to stop using this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
SKYRIZI is a clear and colourless to slightly yellow liquid in a pre-filled syringe with needle guard.
Ingredients
SKYRIZI contains 75 mg of risankizumab as the active ingredient in 0.83 mL solution in each pre-filled syringe.
The other ingredients include:
- Sodium succinate hexahydrate
- Succinic acid
- Sorbitol
- Polysorbate 20
- Water for injections
Distributor
SKYRIZI is distributed in Australia by:
AbbVie Pty Ltd
241 O'Riordan Street
Mascot NSW 2020
Australia
SKYRIZI is distributed in New Zealand by:
AbbVie Limited
6th Floor, 156 158 Victoria Street
Wellington 6011
New Zealand
This leaflet was prepared in October 2019
AUST R 304226
Version 02
Published by MIMS December 2019
 3. Prior to the start of the intravenous infusion, the content of the infusion bag or glass bottle should be at room temperature. The utilisation of a 0.2 micrometre infusion in-line filter is not mandatory.
3. Prior to the start of the intravenous infusion, the content of the infusion bag or glass bottle should be at room temperature. The utilisation of a 0.2 micrometre infusion in-line filter is not mandatory.
 Adverse reactions reported in > 3% of subjects in the maintenance study and at a higher rate than placebo are shown in Table 4.
Adverse reactions reported in > 3% of subjects in the maintenance study and at a higher rate than placebo are shown in Table 4.


 Examination of age, gender, race, body weight, baseline PASI score, concurrent psoriatic arthritis, previous non-biologic systemic treatment, previous biologic treatment, and previous failure of a biologic did not identify differences in response to Skyrizi among these subgroups.
Examination of age, gender, race, body weight, baseline PASI score, concurrent psoriatic arthritis, previous non-biologic systemic treatment, previous biologic treatment, and previous failure of a biologic did not identify differences in response to Skyrizi among these subgroups. For subjects who had PASI 50 to < PASI 90 with adalimumab at Week 16 and were re-randomised, differences in PASI 90 response rates between switching to Skyrizi and continuing adalimumab were noted as early as 4 weeks after re-randomisation (49.1% vs 26.8%, respectively). 66.0% (35/53) of subjects achieved PASI 90 following 28 weeks of Skyrizi, compared with 21.4% (12/56) who continued to receive adalimumab. Other levels of response were also higher following Skyrizi: 39.6% PASI 100, 39.6% sPGA of clear, and 73.6% sPGA of clear or almost clear had response after switching to Skyrizi, compared with 7.1% PASI 100, 7.1% sPGA of clear, and 33.9% sPGA of clear or almost clear who continued to receive adalimumab.
For subjects who had PASI 50 to < PASI 90 with adalimumab at Week 16 and were re-randomised, differences in PASI 90 response rates between switching to Skyrizi and continuing adalimumab were noted as early as 4 weeks after re-randomisation (49.1% vs 26.8%, respectively). 66.0% (35/53) of subjects achieved PASI 90 following 28 weeks of Skyrizi, compared with 21.4% (12/56) who continued to receive adalimumab. Other levels of response were also higher following Skyrizi: 39.6% PASI 100, 39.6% sPGA of clear, and 73.6% sPGA of clear or almost clear had response after switching to Skyrizi, compared with 7.1% PASI 100, 7.1% sPGA of clear, and 33.9% sPGA of clear or almost clear who continued to receive adalimumab.
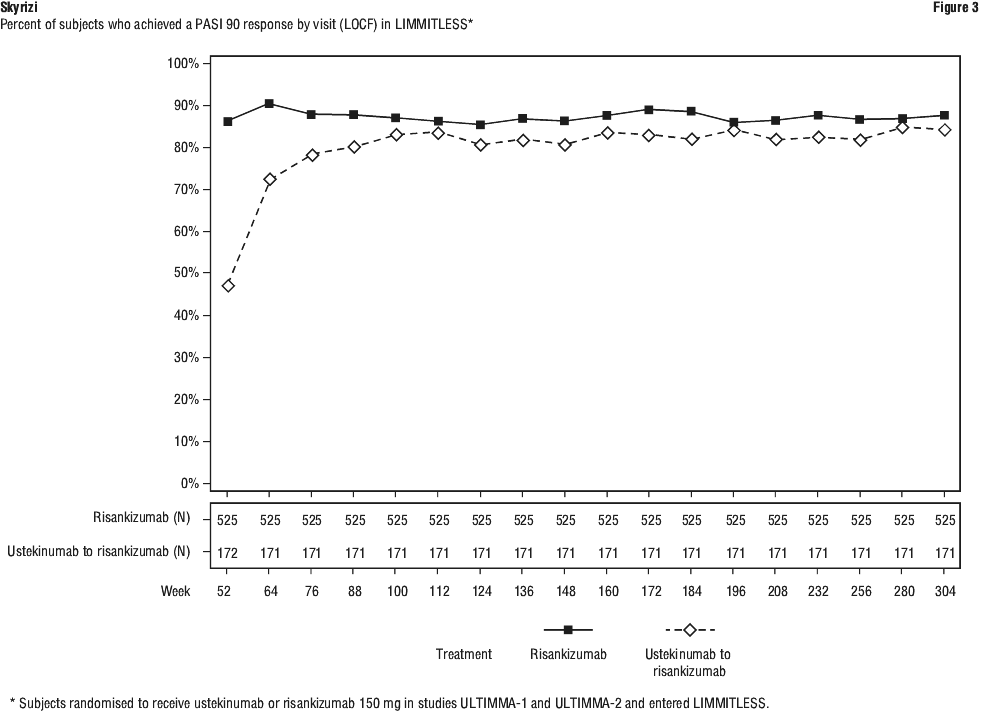
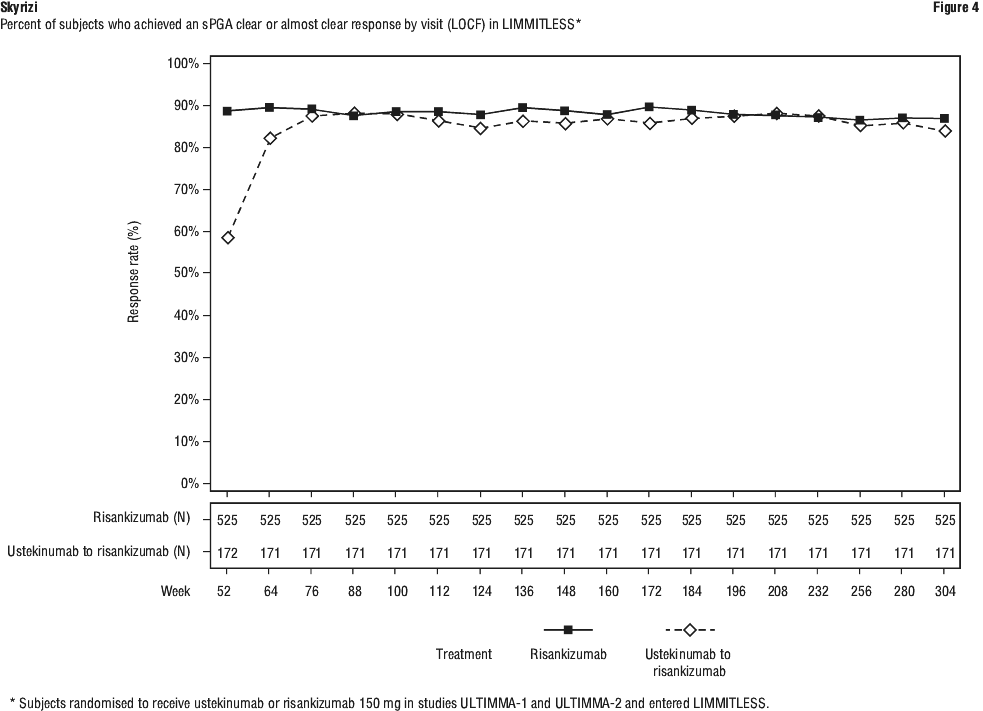
 In ULTIMMA-1 and ULTIMMA-2, significantly greater improvements in psoriasis symptoms (itch, pain, redness and burning, as measured by the Psoriasis Symptom Score [PSS]) were demonstrated with Skyrizi compared with placebo at Week 16. A significantly greater proportion of subjects on Skyrizi achieved a PSS of 0 (symptom-free) at Week 16 compared with ustekinumab and with placebo. By Week 52, 55.7% (333/598) of subjects on Skyrizi reported no itch, pain, redness or burning.
In ULTIMMA-1 and ULTIMMA-2, significantly greater improvements in psoriasis symptoms (itch, pain, redness and burning, as measured by the Psoriasis Symptom Score [PSS]) were demonstrated with Skyrizi compared with placebo at Week 16. A significantly greater proportion of subjects on Skyrizi achieved a PSS of 0 (symptom-free) at Week 16 compared with ustekinumab and with placebo. By Week 52, 55.7% (333/598) of subjects on Skyrizi reported no itch, pain, redness or burning. The percent of subjects achieving ACR20 responses in study KEEPsAKE1 through Week 24 is shown in Figure 5.
The percent of subjects achieving ACR20 responses in study KEEPsAKE1 through Week 24 is shown in Figure 5. In both studies, improvements were shown in all components of the ACR scores including subject's assessment of pain (see Table 10). These responses were maintained through Week 52.
In both studies, improvements were shown in all components of the ACR scores including subject's assessment of pain (see Table 10). These responses were maintained through Week 52. Treatment with Skyrizi resulted in statistically significant improvement in the skin manifestations of psoriasis in subjects with psoriatic arthritis.
Treatment with Skyrizi resulted in statistically significant improvement in the skin manifestations of psoriasis in subjects with psoriatic arthritis.
 At Week 4, a higher proportion of subjects treated with Skyrizi achieved a CDAI < 150 compared to placebo (ADVANCE, Skyrizi = 18%, placebo = 10%, p ≤ 0.05; MOTIVATE, Skyrizi = 21%, placebo = 11%, p ≤ 0.01).
At Week 4, a higher proportion of subjects treated with Skyrizi achieved a CDAI < 150 compared to placebo (ADVANCE, Skyrizi = 18%, placebo = 10%, p ≤ 0.05; MOTIVATE, Skyrizi = 21%, placebo = 11%, p ≤ 0.01). In ADVANCE, a higher proportion of subjects treated with Skyrizi with and without prior biologic failure achieved CDAI < 150 compared to placebo (with prior biologic failure, Skyrizi = 42%, placebo = 26%; Without prior biologic failure, Skyrizi = 49%, placebo = 23%).
In ADVANCE, a higher proportion of subjects treated with Skyrizi with and without prior biologic failure achieved CDAI < 150 compared to placebo (with prior biologic failure, Skyrizi = 42%, placebo = 26%; Without prior biologic failure, Skyrizi = 49%, placebo = 23%). Deep remission at Week 52 was observed at higher rates in subjects treated with Skyrizi I.V./Skyrizi S.C. compared to subjects who received Skyrizi I.V./placebo S.C. (28% vs. 10%, respectively, p < 0.001).
Deep remission at Week 52 was observed at higher rates in subjects treated with Skyrizi I.V./Skyrizi S.C. compared to subjects who received Skyrizi I.V./placebo S.C. (28% vs. 10%, respectively, p < 0.001).
 A significantly greater proportion of subjects treated with Skyrizi compared to placebo had no abdominal pain (36% vs 26%, respectively, p < 0.01) and no bowel urgency (44% vs 28%, respectively, p < 0.00001) at Week 12.
A significantly greater proportion of subjects treated with Skyrizi compared to placebo had no abdominal pain (36% vs 26%, respectively, p < 0.01) and no bowel urgency (44% vs 28%, respectively, p < 0.00001) at Week 12.
