What is in this leaflet
This leaflet answers some common questions about Soliris. It does not contain all the available information.
It does not take the place of talking to your doctor, nurse or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you having Soliris against the benefits they expect it will have for you.
If you have any concerns about this medicine, ask your doctor or nurse.
Keep this leaflet. You may need to read it again.
What Soliris is used for
Soliris is a medicine containing an active substance called eculizumab rmc which belongs to a class of medicines called monoclonal antibodies.
Soliris is used for the treatment of patients with a disease that affects red blood cells called Paroxysmal Nocturnal Haemoglobinuria (PNH).
Soliris is also used to treat patients with a condition called atypical Haemolytic Uraemic Syndrome (aHUS).
How it works
Patients with PNH lack naturally occurring protective proteins on the surface of some of their red blood cells. In unaffected individuals, these proteins protect red blood cells from destruction by the body’s inflammatory response. PNH patients lack these protective proteins and their red blood cells can be destroyed. This can lead to low red blood cell counts (anaemia), tiredness, difficulty in functioning, pain, dark urine, kidney failure, shortness of breath and blood clots.
Soliris can block the body’s inflammatory response, and its ability to attack and destroy blood cells. In this way Soliris improves anaemia, fatigue, and other signs and symptoms of PNH.
Patients with aHUS have an inflammatory condition which affects the blood system and kidney. This can lead to reduced or lost function of the kidneys or other organs, low blood counts (low platelets and anaemia), tiredness and difficulty functioning. Soliris works by blocking the body’s inflammatory response and its ability to attack and destroy its own vulnerable blood and kidney cells.
Before you are given Soliris
When you must not be given Soliris
Soliris treatment may reduce your natural resistance to infections, especially against certain bacteria that can cause meningococcal meningitis (severe infection of the lining of the brain) and sepsis (infection in the blood), as well as other infections caused by similar bacteria (e.g. widespread gonorrhoea).
DO NOT use Soliris if:
- you have not been vaccinated against Neisseria meningitidis, a bacteria that causes meningococcal infection,
- you are not up to date with your meningococcal vaccination,
- if it is less than 2 weeks after receiving your meningococcal vaccination and you are not taking antibiotics to reduce the risk of infection,
- you have meningitis
Do not use Soliris if you have had an allergic reaction to:
- Soliris, or any of the ingredients listed at the end of this leaflet, or
- any other proteins of mouse origin
Symptoms of an allergic reaction may include;
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
If you are not sure whether you should be treated with Soliris, talk to your doctor or nurse.
Before you start Soliris treatment
You must be aware of the following signs and symptoms of a meningococcal infection:
- headache with nausea or vomiting
- headache with a stiff neck or stiff back
- fever
- rash
- confusion
- severe muscle aches with flu-like symptoms
- sensitivity to light
Call your doctor immediately and go to Accident and Emergency at your nearest hospital if you have any of the symptoms listed above.
Patient Safety Information Card
Because of the importance of rapidly identifying and treating certain types of infection you will be provided with a Patient Safety Information Card.
You must carry this card with you at all times and show it to any doctor or nurse that treats you.
You must receive a meningococcal vaccine at least 2 weeks before your first dose of Soliris unless you have already been vaccinated with this vaccine or you take antibiotics to reduce the risk of infection until 2 weeks after you have been vaccinated.
If you have been vaccinated with a meningococcal vaccine in the past, you might need a booster dose before or at the time of starting Soliris. Your doctor will decide if you need another dose of a meningococcal vaccine.
You should also be aware that vaccination may not prevent this type of infection. You may need antibiotics to prevent infection.
If you are at risk of gonorrhoea, ask your doctor or nurse for advice before using this medicine.
If you are less than 18 years old, you must also be vaccinated against Haemophilus influenzae and pneumococcal infections. Your doctor will arrange this according to the national vaccination recommendations for your age group.
Tell your doctor if you have an infection. Soliris may reduce your natural resistance to infection.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Allergic or anaphylactic (more severe allergic) reactions may occur with Soliris treatment. Your doctor or nurse will check for side effects during your infusion and for one hour afterwards. See "Side Effects" for symptoms to look out for.
Tell your doctor if you are pregnant or want to become pregnant. Data in a limited number of pregnant women treated with Soliris do not indicate that Soliris increases the risk of harm to the unborn baby; however, your doctor will discuss the risks and benefits with you. Unless you are planning to become pregnant, you should use adequate contraception whilst being treated with Soliris (and up to 5 months after discontinuing treatment).
Tell your doctor if you are breastfeeding. It is not known whether Soliris passes into breast milk; however, your doctor will discuss the risks and benefits for you and your child if you breastfeed whilst on Soliris treatment.
Tell your doctor if you are on a sodium controlled diet. Soliris contains 115mg sodium per vial, which may need to be considered in calculating your sodium intake.
If you have not told your doctor or nurse about any of the above, tell them before you are given Soliris.
Taking other medicines
Tell your doctor or nurse if you are taking any other medicines, including any that you buy without prescription from your pharmacy, supermarket or health food shop.
The effect of using Soliris on other medicines has not been studied. Ask your doctor or nurse if you have any questions.
How Soliris is given
Follow all directions given to you by your doctor or nurse carefully. They may differ from the information contained in this leaflet.
Soliris will be given to you directly into the vein (intravenously) by a doctor or nurse. Each infusion will take approximately 25 - 45 minutes in adults, and 1 to 4 hours in paediatric patients.
- For adults, Soliris is given once a week for the first four weeks, on the fifth week, the dose is increased and then Soliris is given every two weeks thereafter.
- For children and adolescents who weigh less than 40kg, Soliris will be given at a frequency and dose that varies depending on their weight
- Children and adolescents who weigh more than 40kg are treated with the adult dosing
- If you are having plasma exchange or plasma infusion you may receive additional doses of Soliris
If you miss a dose
If you forget or miss your appointment for a Soliris infusion, contact your doctor immediately.
If you are given too much (overdose)
There have been no reported overdoses of Soliris. As Soliris is given to you under the supervision of your doctor, it is unlikely that you will receive too much.
While you are using Soliris
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Keep appointments with your doctor or clinic. It is important to have the infusion with Soliris at the appropriate time to make sure the medicine has the best chance of providing treatment for the condition.
Have any tests when your doctor says to. Your doctor may wish to test your body’s response to Soliris or may wish to test your body’s response if you stop therapy.
Things you must not do
Do not stop taking Soliris without checking with your doctor.
If you forget or miss a Soliris infusion, call your doctor immediately.
If you have PNH, stopping treatment with Soliris may cause a sudden and serious breakdown of your red blood cells.
Symptoms or problems from red blood cell breakdown include:
- a large drop in your red blood cell count causing anaemia. Symptoms include tiredness, headaches, being short of breath when exercising, dizziness and looking pale
- confusion or change in how alert you are
- chest pain or angina
- dark urine
- blood clots
If you experience any of these symptoms, contact your doctor immediately.
Your doctor will need to monitor you closely for at least 8 weeks after stopping Soliris.
If you have aHUS, stopping treatment with Soliris may cause small blood clots (known as thrombotic microangiopathy or TMA). Your doctor will discuss the possible side effects with you and explain the risks. Your doctor will want to monitor you closely.
Symptoms or problems from TMA may include:
- low blood platelet count, leading to bruising or bleeding more easily than normal
- a large drop in your red blood cell count causing anaemia. Symptoms include tiredness, headaches, being short of breath when exercising, dizziness and looking pale
- confusion or change in how alert you are
- seizures
- chest pain or angina
- decreased urination (kidney problems)
- shortness of breath
- blood clots
If you experience any of these symptoms, contact your doctor immediately.
Side Effects
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Your doctor has weighed the risks of using this medicine against the benefits they expect it will have for you.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Tell your doctor if you notice any of the following and they worry you:
- headaches
- high fever, chills, confusion, rapid breathing
- dizziness, light headedness or shakes
- runny or stuffy nose and colds
- sore throat or mouth ulcers
- cough
- chest or lung infection
- difficulty sleeping
- high blood pressure
- being short of breath (especially when exercising)
The above list includes the more common side effects of Soliris.
Tell your doctor as soon as possible if you notice any of the following:
- bleeding or bruising more easily than normal
- tiredness or unusual weakness
- flu-like symptoms such as fever, sore throat, cough and chills
- looking pale
- confusion or change in how alert you are
- difficulty breathing
- chest tightness
- chest pain or angina
- fast or irregular heartbeats, also called palpitations
- dark urine or blood in the urine
- difficulties or pain when urinating
- diarrhoea
- nausea or vomiting
- stomach pain or discomfort
- constipation
- indigestion or an uncomfortable feeling in the stomach or belching after eating
- rash or itchy skin
- hair loss
- joint pain
- back or neck pain
- muscle spasms or aching muscles, muscle tenderness or weakness not caused by exercise
- loss of appetite
- loss of taste
- pins and needles in your hands or feet
- depression, anxiety or mood swings
- abnormal dreams
Tell your doctor or nurse immediately if you experience any side effects during or after your Soliris infusion.
Allergic reactions are not common, but they may be serious. Your doctor or nurse will monitor you for one hour after your infusion.
Tell your doctor as soon as possible if you notice signs of an infection.
Examples of infection include;
- Sinus, throat or lung infection (e.g. bronchitis or pneumonia)
- cold sores (herpes simplex)
- urinary tract infection (UTI) or cystitis
- gastro or stomach flu
- gum or tooth infections
- viral infection
Soliris may increase your susceptibility to infection. Some infections are serious and can be life-threatening. You may need urgent medical attention.
Tell your doctor immediately and go to Accident and Emergency at your nearest hospital if you experience any of the following symptoms:
- headache with nausea or vomiting
- headache with a stiff neck or stiff back
- fever
- rash
- confusion
- severe muscle aches with flu-like symptoms,
- sensitivity to light
These are possible symptoms of meningococcal infection. If you have meningococcal infection you need urgent medical attention.
Always carry your Soliris Patient Safety Information Card which lists the symptoms of meningococcal disease and important contact information.
If you get any side effects, do not stop Soliris without first talking to your doctor.
Ask your doctor or nurse to answer any questions you may have.
Storing Soliris
Soliris will be stored in refrigerated conditions (2°C to 8°C) in the hospital or pharmacy.
Soliris vials in the original package may be removed from refrigerated storage (up to 25°C) for only one single period of up to 3 days. At the end of this period unopened product can be put back in the refrigerator.
Product Description
What Soliris looks like
Soliris is a clear, colourless, solution contained in a 30mL glass vial.
Ingredients
Active ingredient
- eculizumab rmc
Other ingredients
- sodium chloride
- monobasic sodium phosphate monohydrate
- dibasic sodium phosphate heptahydrate
- polysorbate 80
- water for injections
Manufacturer/Supplier
Alexion Pharmaceuticals Australasia Pty Ltd
Suite 401, Level 4, Building A.
20 Rodborough Rd. Frenchs Forest.
NSW 2086.
Medical enquiries: 1800 788 189
AUST R 138885
This leaflet was revised in June 2020.
Published by MIMS August 2020
 The safety and efficacy of Soliris for the treatment of NMOSD in paediatric patients below the age of 18 years have not been established.
The safety and efficacy of Soliris for the treatment of NMOSD in paediatric patients below the age of 18 years have not been established.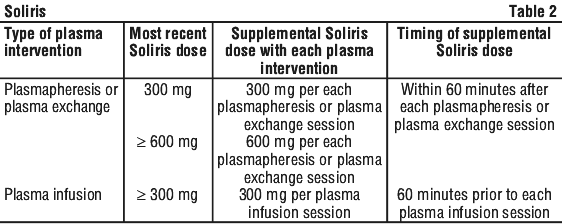

 In the placebo controlled clinical study, serious adverse reactions occurred among 4 (9%) patients receiving Soliris and 9 (21%) patients receiving placebo. The serious reactions included infections and progression of PNH. No deaths occurred in the study and no patients receiving Soliris experienced a thrombotic event; one thrombotic event occurred in a patient receiving placebo.
In the placebo controlled clinical study, serious adverse reactions occurred among 4 (9%) patients receiving Soliris and 9 (21%) patients receiving placebo. The serious reactions included infections and progression of PNH. No deaths occurred in the study and no patients receiving Soliris experienced a thrombotic event; one thrombotic event occurred in a patient receiving placebo. In studies C08-002A/B, C08-003A/B and C10-004 combined, 60% (47/78) of patients experienced a serious adverse event (SAE).
In studies C08-002A/B, C08-003A/B and C10-004 combined, 60% (47/78) of patients experienced a serious adverse event (SAE).
 Analysis of retrospectively collected adverse event data from paediatric and adult patients enrolled in aHUS C09-001r revealed a safety profile that was similar to that which was observed in the prospective studies. C09-001r included 19 paediatric patients less than 18 years of age.
Analysis of retrospectively collected adverse event data from paediatric and adult patients enrolled in aHUS C09-001r revealed a safety profile that was similar to that which was observed in the prospective studies. C09-001r included 19 paediatric patients less than 18 years of age.


 In TRIUMPH, patients treated with Soliris had significantly reduced (p < 0.001) haemolysis resulting in improvements in anaemia as indicated by increased haemoglobin stabilization and reduced need for RBC transfusions compared to placebo-treated patients (see Table 12). These effects were seen among patients within each of the three pre-study RBC transfusion strata (4-14 units; 15-25 units; > 25 units). After 3 weeks of Soliris treatment, patients reported less fatigue and improved health related quality of life.
In TRIUMPH, patients treated with Soliris had significantly reduced (p < 0.001) haemolysis resulting in improvements in anaemia as indicated by increased haemoglobin stabilization and reduced need for RBC transfusions compared to placebo-treated patients (see Table 12). These effects were seen among patients within each of the three pre-study RBC transfusion strata (4-14 units; 15-25 units; > 25 units). After 3 weeks of Soliris treatment, patients reported less fatigue and improved health related quality of life.


 Patients in study C08-002A/B received Soliris for a minimum of 26 weeks. After completion of the initial 26 week treatment period, most patients continued to receive Soliris by enrolling into an extension study. The median duration of Soliris therapy in study C08-002A/B was approximately 100 weeks (range: 2 to 186 weeks).
Patients in study C08-002A/B received Soliris for a minimum of 26 weeks. After completion of the initial 26 week treatment period, most patients continued to receive Soliris by enrolling into an extension study. The median duration of Soliris therapy in study C08-002A/B was approximately 100 weeks (range: 2 to 186 weeks).
 Patients in study C10-004 received Soliris for a minimum of 26 weeks. After completion of the initial 26 week treatment period, most patients elected to continue on chronic dosing.
Patients in study C10-004 received Soliris for a minimum of 26 weeks. After completion of the initial 26 week treatment period, most patients elected to continue on chronic dosing. Longer-term treatment with Soliris (median 52 weeks ranging from 15 to 126 weeks) was associated with an increased rate of clinically meaningful improvements. When Soliris treatment was continued for more than 26 weeks, 3 additional patients (63% of patients in total) achieved complete TMA response and 4 additional patients (98% of patients in total) achieved haematologic normalization. At the last evaluation, 25 of 41 patients (61%) achieved eGFR improvement of ≥ 15 mL/min/1.73 m2 from baseline.
Longer-term treatment with Soliris (median 52 weeks ranging from 15 to 126 weeks) was associated with an increased rate of clinically meaningful improvements. When Soliris treatment was continued for more than 26 weeks, 3 additional patients (63% of patients in total) achieved complete TMA response and 4 additional patients (98% of patients in total) achieved haematologic normalization. At the last evaluation, 25 of 41 patients (61%) achieved eGFR improvement of ≥ 15 mL/min/1.73 m2 from baseline. The efficacy of Soliris for the treatment of NMOSD was established in Study ECU-NMO-301, a randomised, double-blind, placebo-controlled event driven trial that enrolled 143 patients with NMOSD who were anti-AQP4 antibody positive and met the following criteria at screening:
The efficacy of Soliris for the treatment of NMOSD was established in Study ECU-NMO-301, a randomised, double-blind, placebo-controlled event driven trial that enrolled 143 patients with NMOSD who were anti-AQP4 antibody positive and met the following criteria at screening: Baseline characteristics were similar between treatment groups, including mean age at first dose (43.9 in the Soliris group, and 45.0 in the placebo group), and gender [91.7% female (Soliris) versus 89.4% female (placebo)].
Baseline characteristics were similar between treatment groups, including mean age at first dose (43.9 in the Soliris group, and 45.0 in the placebo group), and gender [91.7% female (Soliris) versus 89.4% female (placebo)]. The primary endpoint for Study ECU-NMO-301 was the time-to-first on-trial relapse as adjudicated by an independent committee who were blinded to treatment. On-trial relapse was defined as a new onset of neurologic symptoms or worsening of existing neurologic symptoms with an objective change (i.e. clinical sign) on neurologic examination that persisted for more than 24 hours as confirmed by the treating physician who was blinded to treatment. An adjudicated on-trial relapse was defined as an on-trial relapse that was positively adjudicated by the Relapse Adjudication Committee. A key secondary endpoint was the adjudicated on-trial Annualised Relapse Rate (ARR), which was computed for each patient as the number of relapses divided by the time in years.
The primary endpoint for Study ECU-NMO-301 was the time-to-first on-trial relapse as adjudicated by an independent committee who were blinded to treatment. On-trial relapse was defined as a new onset of neurologic symptoms or worsening of existing neurologic symptoms with an objective change (i.e. clinical sign) on neurologic examination that persisted for more than 24 hours as confirmed by the treating physician who was blinded to treatment. An adjudicated on-trial relapse was defined as an on-trial relapse that was positively adjudicated by the Relapse Adjudication Committee. A key secondary endpoint was the adjudicated on-trial Annualised Relapse Rate (ARR), which was computed for each patient as the number of relapses divided by the time in years. The treatment effect on the time-to first-adjudicated on-trial relapse was observed in patients across all IST subgroups, including the subgroup with no ISTs at Baseline. During the treatment phase of the trial, 76% of patients received concomitant IST, including chronic corticosteroids; 24% of patients did not receive concomitant IST or chronic corticosteroids during the treatment phase of the trial.
The treatment effect on the time-to first-adjudicated on-trial relapse was observed in patients across all IST subgroups, including the subgroup with no ISTs at Baseline. During the treatment phase of the trial, 76% of patients received concomitant IST, including chronic corticosteroids; 24% of patients did not receive concomitant IST or chronic corticosteroids during the treatment phase of the trial. The annualised rate of on-trial relapse associated hospitalisation was 0.04 for Soliris versus 0.31 for placebo. The annualised rate of on-trial relapse associated acute relapse treatment was 0.07 for Soliris versus 0.42 for placebo (IV methylprednisolone) and 0.02 for Soliris versus 0.19 for placebo (plasma exchange).
The annualised rate of on-trial relapse associated hospitalisation was 0.04 for Soliris versus 0.31 for placebo. The annualised rate of on-trial relapse associated acute relapse treatment was 0.07 for Soliris versus 0.42 for placebo (IV methylprednisolone) and 0.02 for Soliris versus 0.19 for placebo (plasma exchange).

 Patients in study C10-003 received Soliris for a minimum of 26 weeks. After completion of the initial 26 week treatment period, most patients elected to continue on chronic dosing. Reduction in terminal complement activity was observed in all patients after commencement of Soliris. Soliris reduced signs of complement mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks.
Patients in study C10-003 received Soliris for a minimum of 26 weeks. After completion of the initial 26 week treatment period, most patients elected to continue on chronic dosing. Reduction in terminal complement activity was observed in all patients after commencement of Soliris. Soliris reduced signs of complement mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. Longer-term treatment with Soliris (median 55 weeks; ranging from 1 day to 107 weeks) was associated with an increased rate of clinically meaningful improvements. When Soliris treatment was continued for more than 26 weeks, 1 additional patient (68% of patients in total) achieved complete TMA response and 2 additional patients (91% of patients in total) achieved haematologic normalization. At the last evaluation, 19 of 22 patients (86%) achieved eGFR improvement of ≥ 15 mL/min/1.73 m2 from baseline. No patient required new dialysis with Soliris.
Longer-term treatment with Soliris (median 55 weeks; ranging from 1 day to 107 weeks) was associated with an increased rate of clinically meaningful improvements. When Soliris treatment was continued for more than 26 weeks, 1 additional patient (68% of patients in total) achieved complete TMA response and 2 additional patients (91% of patients in total) achieved haematologic normalization. At the last evaluation, 19 of 22 patients (86%) achieved eGFR improvement of ≥ 15 mL/min/1.73 m2 from baseline. No patient required new dialysis with Soliris.