
SUMMARY CMI
STEGLATRO®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about taking this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being taken differently. Please report side effects. See the full CMI for further details.
1. Why am I taking STEGLATRO?
STEGLATRO contains the active ingredient ertugliflozin. STEGLATRO can be used to lower your blood sugar (glucose) alone or in combination with certain other medicines, along with a recommended diet and exercise program.
For more information, see Section 1. Why am I taking STEGLATRO? in the full CMI.
2. What should I know before I take STEGLATRO?
Do not use if you have ever had an allergic reaction to ertugliflozin or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I take STEGLATRO? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with STEGLATRO and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take STEGLATRO?
- Take one tablet once a day.
More instructions can be found in Section 4. How do I take STEGLATRO? in the full CMI.
5. What should I know while taking STEGLATRO?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while taking STEGLATRO? in the full CMI.
6. Are there any side effects?
Less serious side effects include yeast infections of the vagina or penis, changes in urination, or low blood sugar if you take STEGLATRO with insulin or certain other diabetes medicines.
Serious side effects include dehydration (losing too much water from your body), ketoacidosis (increased ketones in your blood or urine), genital infection or urinary tract infection.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
FULL CMI
STEGLATRO®
Active ingredient: Ertugliflozin pyroglutamic acid
Consumer Medicine Information (CMI)
This leaflet provides important information about taking STEGLATRO. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about taking STEGLATRO.
Where to find information in this leaflet:
1. Why am I taking STEGLATRO?
2. What should I know before I take STEGLATRO?
3. What if I am taking other medicines?
4. How do I take STEGLATRO?
5. What should I know while taking STEGLATRO?
6. Are there any side effects?
7. Product details
1. Why am I taking STEGLATRO?
STEGLATRO contains the active ingredient ertugliflozin. STEGLATRO is a medicine called a sodium-glucose co-transporter 2 (SGLT2) inhibitor that lowers blood sugar levels in people with type 2 diabetes mellitus. STEGLATRO helps remove sugar from the body through urination. STEGLATRO by itself is unlikely to cause low blood sugar because it does not work when your blood sugar is low.
STEGLATRO can be used to lower your blood sugar (glucose) alone or in combination with certain other medicines, along with a recommended diet and exercise program.
2. What should I know before I take STEGLATRO?
Warnings
Do not take STEGLATRO if:
- you are allergic to ertugliflozin, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can take this medicine.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin or you may feel faint.
- you have poorly functioning kidneys since STEGLATRO requires good functioning kidneys to work well
Tell your doctor if you:
- have type 1 diabetes
- have or have had increased ketones in the blood or urine (diabetic ketoacidosis)
- are going to have surgery
- are eating less due to illness, surgery, or a change in your diet
- drink alcohol very often, or drink a lot of alcohol in the short term (“binge” drinking)
- have kidney problems
- have liver problems, because STEGLATRO is not recommended for patients with severe liver disease
- take other diabetes medicines, you are more likely to get low blood sugar with certain medicines
- have or have had yeast infections of the vagina or penis
- take any medicines for any other condition
- have allergies to any other medicines or other substances such as foods, preservatives or dyes
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Talk to your doctor if you are pregnant or intend to become pregnant. It is not known if STEGLATRO may harm your unborn baby. If you are pregnant, talk with your doctor about the best way to control your blood sugar while you are pregnant.
Do not use STEGLATRO if you are pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed. It is not known if STEGLATRO passes into breast milk. Talk with your doctor about the best way to feed your baby if you take STEGLATRO. Do not use STEGLATRO if you are breast-feeding or plan to breast-feed.
Children
It is not known if STEGLATRO is safe and effective in children under 18 years of age.
Elderly
In studies, STEGLATRO worked well in and was generally well-tolerated by older patients. People 65 years or older were more likely to get dehydrated while taking STEGLATRO compared to younger patients. No dose adjustment is necessary based on age.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or herbal supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
STEGLATRO may be taken with most medicines.
Some medicines may interfere with STEGLATRO and affect how it works.
Be sure to tell your doctor if you are taking water pills (diuretics), as you may be more likely to get dehydrated. See Section 6. Are there any side effects?
When you take STEGLATRO with certain other diabetes medicines, you are more likely to get low blood sugar. See Section 6. Are there any side effects?
Tell your doctor or pharmacist if you are taking lithium because STEGLATRO can lower the amount of lithium in your blood.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect STEGLATRO.
4. How do I take STEGLATRO?
How much to take
- Take one tablet once a day. Your doctor will decide the dose of STEGLATRO suitable for you.
- Take STEGLATRO until your doctor tells you to stop.
When to take STEGLATRO
- Take STEGLATRO at the same time every morning. STEGLATRO can be taken with or without food.
If you forget to take STEGLATRO
STEGLATRO should be taken regularly at the same time each day. If you miss your dose at the usual time, take it as soon as you remember.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take 2 doses of STEGLATRO on the same day.
If you take too much STEGLATRO
If you think that you have taken too much STEGLATRO, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while taking STEGLATRO?
Things you should do
- If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking STEGLATRO.
- If you become pregnant while taking STEGLATRO, tell your doctor immediately.
- Continue to take STEGLATRO for as long as your doctor prescribes it so that you can continue to help control your blood sugar. This medicine helps to control your condition but will not cure it. It is important to keep taking your medicine even if you feel well.
Call your doctor straight away if:
- your body is under some types of stress, such as fever, trauma (such as a car accident), infection, or surgery. The amount of diabetes medicine you need may change.
Remind any doctor, dentist or pharmacist you visit that you are taking STEGLATRO.
Things you should not do
- Do not stop taking this medicine suddenly or lower the dosage without checking with your doctor.
- Do not take STEGLATRO to treat any other complaints unless your doctor tells you to.
- Do not give STEGLATRO to anyone else, even if they have the same condition as you. Remember that your doctor has prescribed this medicine only for you.
Footcare
- Check your feet regularly and see your doctor if you notice any problems. Follow any other advice regarding foot care given by your doctor.
Blood tests
- Your doctor may do blood tests before you start STEGLATRO and while you take it. These tests look to see if your blood sugar level is normal at that moment and how well you have managed your blood sugar over time (called haemoglobin A1c). Blood tests may show changes related to kidney function or high levels of bad cholesterol. Your doctor may change your dose of STEGLATRO based on the results
Driving or using machines
Be careful before you drive or use any machines or tools until you know how STEGLATRO affects you.
STEGLATRO has no or negligible influence on the ability to drive and use machines.
- Do not drive or use any tools or machines if you feel dizzy while taking STEGLATRO.
Taking this medicine in combination with insulin or medicines called insulin secretagogues can cause blood sugar levels to drop too low, which may cause symptoms such as shaking, sweating and change in vision, and may affect your ability to drive and use machines.
Drinking alcohol
Tell your doctor if you drink alcohol very often or drink a lot of alcohol in the short term ("binge" drinking).
Looking after your medicine
- Keep STEGLATRO in its original packaging in a cool dry place where the temperature stays below 30°C.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on windowsills.
Do not take STEGLATRO if the packaging is torn or shows signs of tampering.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to take this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not take this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Yeast infections of the vagina or penis:
If you take STEGLATRO with insulin or certain other diabetes medicines, your blood sugar might get too low. Your doctor might need to lower the dose of your insulin or other diabetes medicine:
| Speak to your doctor if you have any of these side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Dehydration (losing too much water from your body):
You may be more likely to get dehydrated if you:
If possible, check for ketones in your urine, even if your blood sugar is less than 14.0 mmol/L. You may need to be treated in a hospital. Genital infectionThese symptoms could be a sign of a rare but serious life-threatening infection called necrotising fasciitis of the perineum or Fournier's gangrene. Fournier's gangrene must be treated immediately.
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What STEGLATRO contains
| Active ingredient (main ingredient) | Ertugliflozin pyroglutamic acid |
| Other ingredients (inactive ingredients) | Microcrystalline cellulose Lactose monohydrate Sodium starch glycollate Type A Magnesium stearate Hypromellose Macrogol 3350 Triacetin Titanium dioxide Iron oxide red |
STEGLATRO does not contain gluten, sucrose, tartrazine or any other azo dyes.
Do not take this medicine if you are allergic to any of these ingredients.
What STEGLATRO looks like
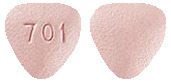
STEGLATRO 5 mg is a pink, triangular-shaped, film-coated tablet marked with '701' on one side and plain on the other side (AUST R: 287619).
STEGLATRO 15 mg is a red, triangular-shaped, film-coated tablet marked with '702' on one side and plain on the other side (AUST R 287622).
Who distributes STEGLATRO
Merck Sharp & Dohme (Australia) Pty Limited
Level 1, Building A, 26 Talavera Road, Macquarie Park NSW 2113
This leaflet was prepared in April 2023.
RCN000025644-AU, RCN000024844-AU
Copyright © 2023 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. All rights reserved.
Published by MIMS July 2023

















 Ertugliflozin pyroglutamic acid is a white to off-white powder that is soluble in ethyl alcohol and acetone, slightly soluble in ethyl acetate and acetonitrile and very slightly soluble in water.
Ertugliflozin pyroglutamic acid is a white to off-white powder that is soluble in ethyl alcohol and acetone, slightly soluble in ethyl acetate and acetonitrile and very slightly soluble in water.