What is in this leaflet
This leaflet answers some common questions about Strensiq. It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using Strensiq against the benefits they expect it will have for you.
If you have any concerns about this medicine, ask your doctor.
Keep this leaflet. You may need to read it again.
What Strensiq is used for
Strensiq is a medicine containing the active substance, asfotase alfa rch.
Strensiq is an enzyme replacement therapy for patients with hypophosphatasia or HPP (diagnosed before the age of 18 years).
How it works
Patients with hypophosphatasia (HPP) have low levels of an enzyme called alkaline phosphatase, which is naturally present in the body and necessary for the proper hardening of bones and teeth. Patients have problems with bone growth and strength, which can lead to broken bones, bone pain, and difficulty walking, as well as difficulties with breathing and a risk of seizures (fits).
Strensiq has been shown to benefit patients’ mineralization of the skeleton and growth.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
This medicine is available only with a doctor’s prescription.
Before you use Strensiq
When you must not use Strensiq
Do not use Strensiq if you have had an allergic reaction to:
- Asfotase alfa rch, or any of the ingredients listed at the end of this leaflet, or
- any other protein of Chinese hamster origin.
Symptoms of an allergic reaction may include;
- shortness of breath, wheezing or difficulty breathing,
- swelling of the face, lips, tongue or other parts of the body,
- rash, itching or hives on the skin.
If you have previously had an allergic reaction to Strensiq, your doctor will discuss with you the next steps and the possibility to restart Strensiq under medical supervision.
Do not use Strensiq after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should be treated with Strensiq, talk to your doctor.
Before you start to use Strensiq
Some known eye-related side-effects have been reported in clinical trials with Strensiq, most likely associated with hypophosphatasia, however, talk to your doctor if you experience any vision problems while being treated with Strensiq.
Early fusion of the bones of the head in children < 5 years of age has been reported in clinical studies of infants with hypophosphatasia, with and without use of Strensiq. Talk to your doctor if you notice any change in the shape of your child’s head.
You may experience a reaction at the injection site (pain, swelling, rash, discolouration) during the injection of Strensiq or during the hours following the injection. If you experience any severe reaction at the injection site, tell your doctor immediately.
Increases of parathyroid hormone (a hormone that controls calcium concentration in your blood) and low calcium levels have been reported in clinical studies with Strensiq. As a consequence, your doctor may ask you to do some blood tests and, depending on the results of the blood test, ask you to take supplements of calcium and oral vitamin D.
Weight gain may occur during your treatment with Strensiq. Your doctor will provide dietary advice as necessary.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you are pregnant or want to become pregnant. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you are breastfeeding. It is not known whether Strensiq passes into breast milk. You should not breast feed while using Strensiq unless you have discussed it with your doctor.
If you have not told your doctor about any of the above, tell them before you start using Strensiq.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without prescription from your pharmacy, supermarket or health food shop.
Some medicines and Strensiq may interfere with each other. They may be affected by Strensiq or may affect how well it works. You may need different amounts of your medicines or may need to take different medicines.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking this medicine.
How to use Strensiq
Follow all directions given to you by your doctor, nurse or pharmacist carefully. They may differ from the information contained in this leaflet.
“How to use Strensiq” will be explained to you by a healthcare professional experienced in the management of patients with metabolic or bone related diseases. After being trained by the doctor or specialized nurse, you can inject Strensiq yourself at home.
Do not inject Strensiq into a vein or muscle. Strensiq is intended for subcutaneous (under the skin) injection.
If you do not understand these instructions, ask your doctor, nurse or pharmacist for help.
How much to use
Your doctor will tell you how much of this medicine (in number of milligrams) you need to inject based on your body weight.
The correct dose will be calculated by your doctor based on a total of 6 milligrams of Strensiq, per kg of body weight, per week.
You will inject either 3 or 6 times a week depending on the recommendation of your doctor.
Your doctor may adjust your dose as your weight changes.
The maximum amount of Strensiq to be injected is 1 mL. If more than 1 mL is required, split the volume equally between two or more syringes, and administer each injection using a separate site.
How to use Strensiq
Please read the following instructions very carefully:
If you are injecting this medicine yourself, you will be shown how to prepare and give the injection by your doctor, pharmacist or nurse.
Do not inject this medicine yourself unless you have received training and you understand the procedure.
You may experience a reaction at the injection site. Please read “Side Effects” very carefully to know what side effects can occur before using this medicine.
When injecting, it is important to change the injection site between different areas of the body to help reduce potential pain and irritation.
Areas with a good amount of fat below the skin (thigh, arm) are the most suitable areas to inject.
How to inject Strensiq:
- Before you begin, take your vial(s) of Strensiq out of the refrigerator. It is recommended to administer the medicine WITHIN 3 HOURS after removing the vial (s) from the refrigerator.
Always use a new vial. Each vial is for single use and should only be punctured once.
- Wash your hands thoroughly with soap and water.
- Place all the items you will need on a clean surface where you will not be disturbed. These should include;
- vial(s) of Strensiq
- syringes and needles
- alcohol swabs (if required)
- gauze or cotton wool
- sharps container
- adhesive bandage
- injection diary (or other recording means).
- Visually check the vial before use. Strensiq should be colourless to slightly yellow in colour. Do not use if it is cloudy, hazy, discoloured or contains particles.
- Remove the protective cap from the vial.
- Remove the plastic cap covering the syringe needle and draw air into the syringe, equal to the volume of medicine to be injected. The volume per injection should not exceed 1mL. If this is the case, multiple injections should be done at different sites.
- Holding the syringe and vial at 45° angle, insert the needle through the sterile rubber top and inject air into the vial.
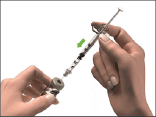
- Turn the vial and syringe upside down. With the needle in the solution, pull the plunger to withdraw the volume of the correct dose into the syringe.
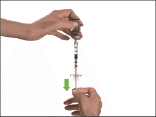
- Before removing the needle from the vial, check the syringe for air bubbles.

If bubbles are present, hold the syringe with the needle pointing upwards and gently tap the side of the syringe until the bubbles float to the top.
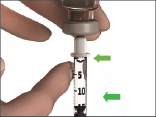
Once all the bubbles are at the top of the syringe, gently push on the plunger to force the bubbles out of the syringe and back into the vial.
After removing the bubbles, check the dose of medicine in the syringe to be sure you have drawn up the correct amount.
Remove the needle from the vial and make sure the needle does not touch anything.
You are now ready to inject the correct dose.
- Choose an injection site. Strensiq should be administered as a subcutaneous injection into the fatty layer just below the skin. This is called the subcutaneous layer and it is just above the muscle. Areas with a substantial amount of fat below the skin (as shown in the diagram) are the most suitable areas to inject.
Your doctor will advise you on the possible injection sites.
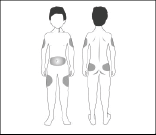
It is important to rotate injection sites, as this may help reduce pain and irritation. Do not inject into areas that are reddened, inflamed, swollen or into any areas in which you feel lumps, firm knots, or pain.
- Using an alcohol-based (isopropyl alcohol or ethanol) solution wipe or clean the site.
- Gently pinch the skin of the chosen injection area between your thumb and index finger.
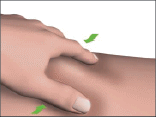
- Holding the syringe like a pencil or a dart, insert the needle into the raised skin so it is at an angle of between 45° and 90° to the skin surface.
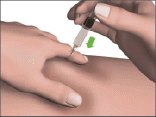
For children, or patients who have little subcutaneous fat or thin skin, a 45° angle may be preferable.
- While continuing to hold the skin, push the syringe plunger to inject the medicine while counting slowly to 10.
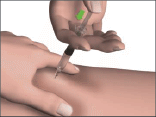
- Remove the needle, release the skin fold and gently place a piece of cotton wool or gauze over the injection site for a few seconds.
This will help seal the punctured tissue and prevent any leakage.
Do not rub the injection site after injection.
- Dispose of your syringe, needle cap and used vial into your sharps container.
DO NOT re-cap the needle.
- Place a small adhesive bandage over the injection site if necessary.
- If you need a second injection for your prescribed dose, get another Strensiq vial and repeat steps 4 through 17.
- Record all details of the injection in your injection diary or using another means. It is recommended that you note:
- where you injected
- the dose injected
- any injection reactions.
If you have any concerns about injection reactions, preparing or administering your injection, discuss them with your doctor or pharmacist.
If you miss a dose
Do not take a double dose to make up for a forgotten dose. Contact your doctor for advice.
If you use too much (overdose)
If you suspect you have accidentally injected a higher dose of Strensiq than prescribed, please contact your doctor for advice or contact the Poisons Information Centre on 13 11 26.
While you are using Strensiq
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
Tell your doctor or pharmacist if you are traveling. You will need to calculate how many vials you will need for your trip. Take extra in case you are away longer than expected.
Discuss traveling with your medicine with your doctor or pharmacist.
If you become pregnant while using Strensiq, tell your doctor immediately. Your doctor will discuss the benefits and risks of continuing treatment with Strensiq during pregnancy, including participation in a monitoring program.
If you intend to breastfeed whilst using STRENSIQ, consult your doctor. Your doctor will discuss the benefits and risks of continuing treatment with Strensiq whilst breastfeeding, including participation in a monitoring program.
If you are having any blood tests, tell your doctor that you are using this medicine. Some medicines can interfere with the results of some tests.
If you need to undergo laboratory tests (giving blood for testing), tell your doctor that you are treated with Strensiq. Strensiq may cause some tests to show wrongly higher or lowerresults. Therefore another type of test may need to be used if you are treated with Strensiq.
Keep all of your doctor’s appointments so that your progress can be checked. Because your dose is calculated based on your weight, it is important that your doctor can monitor your weight and adjust as needed. Your doctor may do some tests to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Side Effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are using Strensiq.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Tell your doctor if you notice any of the following and they worry you:
- reactions at the injection site during the injection of the medicine or during the hours following the injection (which can lead to redness, discolourations, itching, pain and/or swelling)
- chills, fever, rash, irritability,
- skin redness, loose skin, discolouration of the skin, bruising, scarring
- fatty lumps under the surface of the skin or localised loss of fat tissue
- small dent or depression of the skin at the injection site
- pain in hands and feet
- nausea or feeling sick
- numbness of the lips/mouth
- aching muscles, muscle tenderness or weakness not caused by exercise
- hot flush
- infection of the skin at injection site
- headache.
The above list includes the more common side effects of your medicine.
Tell your doctor as soon as possible if you notice any of the following:
- bruising more easily than normal
- eye problems, blurred vision.
Tell your doctor or go to the Emergency Department at your nearest hospital if you notice any of the following;
Strensiq contains a protein and proteins can cause allergic reactions in some people. Symptoms of such reactions include;
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- low blood pressure
- vomiting
- fast heart rate
- choking sensation
The above lists include serious side effects that may require medical attention.
Other side effects not listed above may occur in some patients. Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Storing Strensiq
Keep your medicine in the packaging until it is time to use it. If you take the medicine out of the packaging it may not keep well.
Keep your medicine in the refrigerator (2°C to 8°C). Do not put Strensiq in, or near the freezer compartment, and never inject Strensiq that you know, or suspect, has been frozen.
After opening the vial, the product should be used immediately (within 3 hours maximum at room temperature, up to 25°C)
Keep Strensiq where children cannot reach it.
Disposal
Ask your pharmacist what to do with any medicine that is left over.
Product Description
What Strensiq looks like
Strensiq is a clear, colourless to slightly yellow, solution contained in a glass vial.
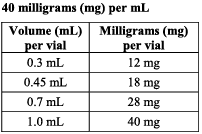
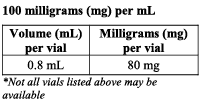
It is available in packs of 12 vials in the following strengths;
Ingredients
Active ingredient
- asfotase alfa rch
Other ingredients
- sodium chloride
- monobasic sodium phosphate monohydrate
- dibasic sodium phosphate heptahydrate
- Water for Injection
Manufacturer
In Australia this product is registered by:
Alexion Pharmaceuticals Australasia Pty Ltd
Suite 401, Level 4, Building A.
20 Rodborough Rd. Frenchs Forest.
NSW 2086.
Medical enquiries: 1800 788 189
Registration numbers;
AUST R 232545
AUST R 232546
AUST R 266984
AUST R 266985
AUST R 266986
This leaflet was prepared in - November 2020
® = Registered Trademark
Published by MIMS December 2020
 Patients should be regularly reviewed for their response to treatment and appropriate dose, including patients who have progressed to adolescence and adulthood.
Patients should be regularly reviewed for their response to treatment and appropriate dose, including patients who have progressed to adolescence and adulthood.
 Baseline characteristics of patients with paediatric onset HPP evaluated in clinical trials included low ALP and one or more of the following; elevated TNSALP biochemical substrates (PPi and PLP), abnormal bone structure (elevated osteoid indices, reduced bone mineral content, skeletal deformities of rickets such as bowed legs, abnormally shaped chest, below normal Z-score for height) or impaired physical function (gross motor weakness, developmental delay, impaired walking, inability to perform activities of daily living). At baseline, patients less than 5 years of age presented with additional morbidities including nephrocalcinosis, seizures, respiratory compromise (including respiratory failure requiring support) and gross motor delays. In Study ENB-002-08/ENB-003-08, most patients (9/11, 81.8%) presented with significant gross motor delays on the BSID-III (Bayley Scales of Infant and Toddler Development, Third Edition) e.g. gross motor scaled scores of 1, which is 3 SDs below the mean SD for healthy age-matched peers. In Study ENB-009-10, 18/19 (94.7%) patients experienced fractures and 18 patients had bone pain severe enough to limit activity.
Baseline characteristics of patients with paediatric onset HPP evaluated in clinical trials included low ALP and one or more of the following; elevated TNSALP biochemical substrates (PPi and PLP), abnormal bone structure (elevated osteoid indices, reduced bone mineral content, skeletal deformities of rickets such as bowed legs, abnormally shaped chest, below normal Z-score for height) or impaired physical function (gross motor weakness, developmental delay, impaired walking, inability to perform activities of daily living). At baseline, patients less than 5 years of age presented with additional morbidities including nephrocalcinosis, seizures, respiratory compromise (including respiratory failure requiring support) and gross motor delays. In Study ENB-002-08/ENB-003-08, most patients (9/11, 81.8%) presented with significant gross motor delays on the BSID-III (Bayley Scales of Infant and Toddler Development, Third Edition) e.g. gross motor scaled scores of 1, which is 3 SDs below the mean SD for healthy age-matched peers. In Study ENB-009-10, 18/19 (94.7%) patients experienced fractures and 18 patients had bone pain severe enough to limit activity.
 Overall survival was improved in the cohort of treated patients with severe perinatal- or infantile-onset HPP, compared to the matched historical control group, with 69/78 (88%) treated patients vs. 13/48 (27%) historical controls alive at last contact. In patients who required any form of respiratory support, 23/29 (79%) of the treated patients survived through their last assessment (median age at last assessment was 3.9 years), versus 1 of 20 (5%) of historical controls.
Overall survival was improved in the cohort of treated patients with severe perinatal- or infantile-onset HPP, compared to the matched historical control group, with 69/78 (88%) treated patients vs. 13/48 (27%) historical controls alive at last contact. In patients who required any form of respiratory support, 23/29 (79%) of the treated patients survived through their last assessment (median age at last assessment was 3.9 years), versus 1 of 20 (5%) of historical controls.


 Based on the results of population pharmacokinetic analysis, body weight was identified to affect asfotase alfa rch clearance and volume of distribution parameters. It is expected that pharmacokinetic exposures will increase with body weight. The impact of immunogenicity on asfotase alfa rch pharmacokinetics varied over time due to the time varying nature of immunogenicity and overall was estimated to decrease pharmacokinetic exposures by less than 20%.
Based on the results of population pharmacokinetic analysis, body weight was identified to affect asfotase alfa rch clearance and volume of distribution parameters. It is expected that pharmacokinetic exposures will increase with body weight. The impact of immunogenicity on asfotase alfa rch pharmacokinetics varied over time due to the time varying nature of immunogenicity and overall was estimated to decrease pharmacokinetic exposures by less than 20%.

