What is in this leaflet
This leaflet answers some common questions about SUBUTEX. It does not contain all the available information.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking SUBUTEX against the benefits you may gain and he/she believes it will help in your treatment.
If you have any concerns about taking SUBUTEX, ask your doctor.
Keep this leaflet. You may want to read it again.
What is SUBUTEX used for?
SUBUTEX is used as part of a medical, social and psychological treatment program for patients dependent on opioids like heroin, morphine, oxycodone or codeine. SUBUTEX is used to help patients overcome this medical condition.
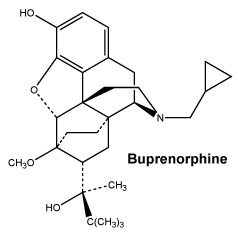
SUBUTEX tablets contain the active ingredient buprenorphine hydrochloride. It acts as a substitute for opioids like heroin, morphine, oxycodone or codeine and it helps withdrawal from opioids over a period of time.
SUBUTEX should be used exactly as prescribed by your doctor.
Ask your doctor if you have any questions about why SUBUTEX has been prescribed for you.
Before you take SUBUTEX
SUBUTEX is not suitable for everyone.
When you must not take SUBUTEX
- If you are under the age of 16 years.
- If you are allergic to buprenorphine or to any of the other ingredients in this medicine (see Product Description below).
- If you have serious breathing problems.
- If you have serious problems with your liver, or if your doctor detects the development of such a problem during treatment.
- If you are intoxicated due to CNS depressant medicines (eg. sedative/hypnotics, narcotic pain killers, anti-anxiety or antipsychotic medicines), alcohol or have delirium tremens (the 'shakes' and hallucinations).
Do not take SUBUTEX if the package is torn, shows signs of tampering or if the tablets do not look quite right.
Before you start to use SUBUTEX
Tell your doctor if you have any of the following before treatment, or develop them during treatment, as your doctor may need to adjust your dose of SUBUTEX:
- if you are pregnant
- if you are breastfeeding
- asthma or other breathing problems;
- thyroid problems;
- prostate problems;
- problems with excess alcohol use;
- problems with drowsiness;
- Addison's disease;
- Kyphoscoliosis (hunchback disease);
- low blood pressure;
- urination problems;
- kidney problems;
- liver problems;
- if you have head injuries or have a condition where you have increased pressure within your head,
- if you have problems related to the biliary tract
- if you have a history of seizures.
- if you have severe mental problems or hallucinations (seeing or hearing things that are not really there)
Some people have died from respiratory failure (inability to breathe) when using benzodiazepines (medicines used to treat anxiety or sleeping problems), or other depressants such as alcohol or other opioids at the same time as SUBUTEX. For further information please discuss with your doctor.
SUBUTEX may cause fatal respiratory failure in children who accidentally ingest it.
Keep this medicine out of reach and sight of children.
SUBUTEX can cause withdrawal symptoms if you take it less than six hours after you use heroin or morphine.
Also, if treatment is stopped abruptly, withdrawal symptoms may occur, which may be delayed in some cases.
SUBUTEX is not intended for occasional use and should be taken only as prescribed.
SUBUTEX may cause drowsiness, which may be made worse if you also drink alcohol or take sedatives or anti-anxiety medicines. If you are drowsy, do not drive or operate machinery.
SUBUTEX may cause your blood pressure to drop suddenly, causing you to feel dizzy if you get up too quickly from sitting or lying down.
Athletes should be aware that this medicine may cause a positive reaction to "anti-doping" tests.
The safety and effectiveness in patients over 65 years of age have not been established.
Your doctor may ask you to have additional blood tests to see if this medication is right for you.
Taking Other Medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop, before you begin treatment with SUBUTEX.
A number of medicines may alter the effects of SUBUTEX. These include:
- certain medicines for treating HIV/AIDS;
- certain medicines for treating fungal and bacterial infections;
- strong pain killers;
- cough medicines containing opioid-related substances;
- certain antidepressants including monoamine oxidase inhibitors;
- certain medicines used to treat fits or epilepsy (anti-convulsants);
- sedating antihistamines;
- sedatives, alcohol;
- anti-anxiety medicines;
- certain medicines for high blood pressure, and
- antipsychotic medicines
- naltrexone.
Tell your doctor if you are scheduled to have surgery using a general anaesthetic.
Do not drink alcohol or take medicines that contain alcohol whilst you are being treated with SUBUTEX. Alcohol and certain other medicines (as listed above) may increase the sedative effects of buprenorphine, which can make driving and operating machinery hazardous.
Some people have died when using sedatives (benzodiazepines) or other depressants, alcohol or other opioids at the same time as SUBUTEX. You should not use benzodiazepines (medicines used to treat anxiety or sleep disorders) whilst you are taking SUBUTEX unless they are prescribed by your doctor.
How to Take SUBUTEX
Do not take SUBUTEX to treat any condition other than the one prescribed for by your doctor.
Do not give SUBUTEX to anyone else, even if their symptoms seem the same as yours. It may harm them.
The tablets are taken sublingually. This means that you place the tablet under your tongue and allow it to dissolve, which may take 2 to 10 minutes. This is the only way the tablets should be taken. Do not swallow or consume food or drink until the tablet is completely dissolved. Do not split or break the tablet.
The tablets will not work if you chew or swallow them whole.
Do not inject SUBUTEX; patients have died from injecting SUBUTEX. Additionally, when injecting SUBUTEX and also taking benzodiazepines (medicines used to treat anxiety or sleeping problems), people were even more likely to die.
How much to take
SUBUTEX is only for adults and children over the age of 16 years. Your doctor will tell you how much SUBUTEX to take and you should always follow medical advice.
On the first day the recommended starting dose is 4-8 mg SUBUTEX with an additional 4 mg depending on your needs as determined by your treating doctor.
- For patients who are still using short acting opioids such as heroin, morphine, oxycodone or codeine: when starting treatment the dose of SUBUTEX should be taken at least 6 hours after your last use of opioids or when the first signs of craving appear
- For patients receiving methadone: before beginning treatment with SUBUTEX, your doctor will probably reduce your dose of methadone to the minimum methadone daily dose that you can tolerate. The first dose of SUBUTEX should be taken at least 24 hours after your last dose of methadone or when the first signs of craving appear.
SUBUTEX may cause withdrawal symptoms if taken while still under the influence of another opioid.
During your treatment, your doctor may increase your dose of SUBUTEX to a maximum of 32mg, depending upon your response to treatment.
After a period of successful treatment, your doctor may gradually reduce your dose.
Depending on your condition, your dose may continue to be reduced under careful medical supervision, until it is stopped altogether.
Do not suddenly stop taking the tablets, as this may cause withdrawal symptoms.
If you miss a dose of SUBUTEX
If you forget to take a dose of SUBUTEX take it as soon as you remember. If you are unsure consult your doctor.
If you take too much of SUBUTEX (overdose)
If you think that you or anyone else may have taken too much SUBUTEX, immediately telephone your doctor or National Poison Centre (in Australia telephone 13 11 26 or in New Zealand telephone 0800 POISON or 0800 764 766), or go to accident and emergency at your nearest hospital. Do this even if there are no signs of discomfort or poisoning.
You may need urgent medical attention.
Keep telephone numbers for these places handy.
If you take too much SUBUTEX, some of the symptoms which may or may not occur are listed in the Side effects section of this leaflet.
Side effects
Like all medicines, SUBUTEX may have unwanted side effects which may need medical treatment.
Ask your doctor or pharmacist to answer any questions you may have.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Many of the common side effects reported with the use of SUBUTEX were related to opioid withdrawal symptoms, such as:
- difficulty sleeping, anxiety, nervousness,
- malaise, fatigue,
- pain in the abdomen, back, joints and muscles, leg cramps, muscle weakness,
- flu-like symptoms such as chills, fever, sore throat, coughing, runny nose, watery eyes and sweating,
- upset stomach and diarrhoea.
Other side effects which have occurred are:
- dry mouth, tooth disorders
- headache, migraine
- sleepiness, dizziness, fainting, vertigo
- abnormal vision
- depression, abnormal thinking, hostility, agitation, paranoid reactions, tremor
- difficulty sleeping
- chest pain, neck pain
- pain in joints, muscles, back, stomach, cramps
- palpitations, flushing
- difficulty in breathing
- yawning, cough, respiratory infection
- nausea, vomiting, constipation, diarrhoea, flatulence or wind, poor appetite, decreased weight
- swelling of the legs and arms, numbness
- fatigue, weakness
- lymph node problems
- hives, rash and itching
- painful periods
- sweating
- difficulty in bladder control
If you think you are suffering from any of the above side-effects, or any other side effects, you should tell your doctor immediately.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital. You may need urgent medical attention.
- There have been rare cases of life-threatening severe hypersensitive reactions with symptoms of severe difficulty in breathing, swelling of the face, lips, mouth or throat.
- Some cases of severe liver problems have occurred during treatment. If you develop severe fatigue, have no appetite or if your skin or eyes look yellow, you have light coloured bowel motions or dark coloured urine, tell your doctor immediately.
Other side-effects not listed above may occur in some patients. Tell your doctor if you notice anything else that is making you feel unwell.
After Using SUBUTEX
If you stop taking SUBUTEX and start using opioids again, you are at risk of being more sensitive to opioids, which could be dangerous. You should talk to your doctor if you start using opioids again.
Presentation and Storage
SUBUTEX is packed in child resistant blisters.
Below are the instructions on how to open the blisters.
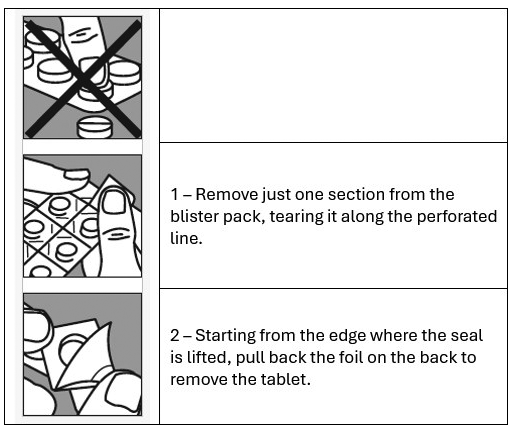
SUBUTEX contains a narcotic that can be a target for people who abuse prescription medicines or street drugs. Therefore, keep your tablets in a safe place to protect them from theft. Keep out of reach and sight of children. Never give them to anyone else.
The tablets should be stored below 30°C in the original package. As with all medicines, keep out of the reach of children. Do not use SUBUTEX after the expiry date that is stamped on the pack.
Product Description
What SUBUTEX Looks Like.
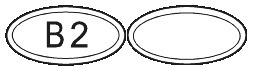
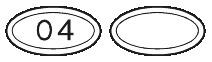
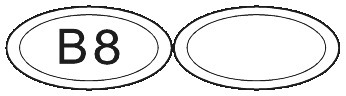
SUBUTEX are white, flat oval shaped tablets. Tablets are debossed with either "04", "B2" or "B8" respectively for SUBUTEX 0.4mg, SUBUTEX 2mg and SUBUTEX 8mg.
Ingredients:
Each SUBUTEX sublingual tablet contains 0.4 mg, 2mg or 8mg buprenorphine (as hydrochloride) as the active ingredient, along with the following inactive ingredients:
lactose monohydrate; mannitol; starch-maize; povidone; citric acid; sodium citrate monohydrate and magnesium stearate.
SUBUTEX:
0.4mg - AUST R 76661
2mg - AUST R 76662
8mg - AUST R 76663
Sponsor:
Indivior Pty Ltd
78 Waterloo Road
Macquarie Park NSW 2113
Australia
For adverse event reporting please contact:
Indivior Pty Ltd
+800-270-81901
[email protected]
Date of most recent amendment: 29 March 2021
Published by MIMS May 2021

 A similar schedule employed Subutex treatment only on the first 5 days (Table 3). The Subutex dose was increased over the first 3 days and then decreased.
A similar schedule employed Subutex treatment only on the first 5 days (Table 3). The Subutex dose was increased over the first 3 days and then decreased.
 In the same study, changes in Cmax and AUClast in subjects with HCV infection without hepatic impairment were not clinically significant in comparison to the healthy subjects.
In the same study, changes in Cmax and AUClast in subjects with HCV infection without hepatic impairment were not clinically significant in comparison to the healthy subjects. The most common adverse events reported were those related to withdrawal symptoms (e.g. abdominal pain, headache, pain, diarrhoea, nausea, muscle aches, anxiety, sweating). In patients with marked opioid dependence, initial administration of buprenorphine can produce a withdrawal effect similar to that associated with naloxone.
The most common adverse events reported were those related to withdrawal symptoms (e.g. abdominal pain, headache, pain, diarrhoea, nausea, muscle aches, anxiety, sweating). In patients with marked opioid dependence, initial administration of buprenorphine can produce a withdrawal effect similar to that associated with naloxone. Compared with intravenous administration, the bioavailability of 0.4 mg and 0.8 mg sublingual buprenorphine tablet doses was 30-35%. With 8 mg sublingual buprenorphine delivered as a solution the buprenorphine bioavailability compared to intravenous administration was 42%.
Compared with intravenous administration, the bioavailability of 0.4 mg and 0.8 mg sublingual buprenorphine tablet doses was 30-35%. With 8 mg sublingual buprenorphine delivered as a solution the buprenorphine bioavailability compared to intravenous administration was 42%.