


What is in this leaflet
This leaflet answers some common questions about SUTENT.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking SUTENT against the benefits it is expected to have for you.
If you have any concerns about this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What SUTENT is used for
SUTENT is used in the treatment of renal cell carcinoma, a type of kidney cancer.
SUTENT is used to treat gastrointestinal stromal tumour (GIST). GIST is a cancer of the stomach and bowels. It is caused by the uncontrolled growth of cells in the wall of the stomach or bowel. SUTENT slows down the growth of these cells.
SUTENT is also used to treat pancreatic neuroendocrine tumours. This is a rare cancer in the cells of the pancreas that release hormones.
Ask your doctor if you have any questions about why SUTENT has been prescribed for you. Your doctor may have prescribed it for another purpose.
SUTENT is only available with a doctor's prescription. It is not addictive.
Use in children
The safety and efficacy of SUTENT have not been established in children.
Before you take SUTENT
When you must not take it
Do not take SUTENT if you have ever had an allergic reaction to sunitinib (the active ingredient in SUTENT) or any of the ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not use SUTENT after the expiry date printed on the pack.
Do not use SUTENT if the packaging shows signs of tampering.
Before you start to take it
You must tell your doctor if:
- you have high blood pressure
- you have or have had an aneurysm (abnormal balloon-like swelling in the wall of an artery)
- you have problems with your heart
- you have or have ever had problems with your liver or kidneys
- you are pregnant or planning to become pregnant.
SUTENT should not be used during pregnancy. Your doctor will discuss the risks with you. - you are breastfeeding.
You should not breastfeed while taking SUTENT.
You should have a dental check up before taking SUTENT.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines that you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines and SUTENT may interfere with each other. Some of these medicines include:
- ketoconazole, a medicine to treat fungal infections
- itraconazole, a medicine to treat fungal infections
- ritonavir, a medicine to treat HIV infection
- erythromycin, a medicine to treat infections
- clarithromycin, a medicine to treat infections
- rifampicin, a medicine to treat tuberculosis and some other infections
- dexamethasone, a medicine to treat dermatitis, asthma and some other conditions
- phenytoin, a medicine to treat seizures
- carbamazepine, a medicine to treat seizures
- phenobarbital (phenobarbitone), a medicine to treat seizures
- St. John's wort (a herbal medicine, also called Hypericum perforatum) to treat anxiety
- medicines used to treat irregular heart beat
- medicines called bisphosphonates, such as zoledronic acid, alendronate pamidronate or ibandronate to treat osteoporosis and some types of cancers
- medicines to treat diabetes.
You may need to take different amounts of your medicines or you may need to use different medicines. Your doctor will advise you.
How to take SUTENT
Follow all directions given to you by your doctor carefully. These directions may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you the dose that you should take. The dose depends on the type of cancer.
For renal cell carcinoma and GIST, the usual dose is 50 mg taken once a day for 4 weeks followed by no medicine for 2 weeks, making a 6-week cycle. Your doctor will let you know how many cycles of treatment you will need.
For pancreatic neuroendocrine tumours, the usual recommended dose is 37.5 mg taken once daily.
Your doctor may change your dose or dosing schedule during treatment.
How to take it
Swallow the capsules with a glass of water.
Take the capsules at about the same time each day. Taking them at the same time each day will help you to remember to take them.
SUTENT can be taken with or without food.
How long to take it
Continue taking SUTENT for as long as your doctor prescribes it.
If you forget to take it
If you miss a dose, do not take an additional dose. Take your usual dose on the next day.
Do not take a double dose to make up for the one that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (Phone Australia 13 11 26 or New Zealand 0800 POISON or 0800 764 766) for advice or go to Accident and Emergency at your nearest hospital, if you think you or anyone else may have taken too much SUTENT. Do this even if there are no signs of discomfort or poisoning.
Keep the telephone numbers for these places handy.
While you are taking SUTENT
Things you must do
Make sure you follow your doctor's instructions and keep all appointments. You will need regular follow-up to make sure the treatment is working.
Your doctor will measure your blood pressure. You will also have blood tests to check for side effects.
Use a proven method of birth control (contraception) to prevent pregnancy while being treated with SUTENT and for at least 4 weeks after finishing treatment with SUTENT.
Tell your doctor immediately if you become pregnant while taking SUTENT.
Tell your doctor if you are going to have surgery, an operation or dental treatment while taking SUTENT.
Tell any doctor, dentist or pharmacist who treats you that you are taking SUTENT.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking SUTENT.
Things you must not do
Do not take SUTENT to treat any other complaints unless your doctor tells you to.
Do not give SUTENT to anyone else, even if their condition seems similar to yours.
Things to be careful of
Avoid drinking grapefruit juice while you are being treated with SUTENT. Grapefruit juice may interact with SUTENT and affect how your body uses this medicine.
Be careful driving, operating machinery or doing jobs that require you to be alert, until you know how SUTENT affects you. SUTENT may make some people feel very tired or dizzy.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking SUTENT.
All medicines can have unwanted side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects. Medicines can affect people in different ways.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following:
- tiredness
- diarrhoea
- nausea (feeling sick) or vomiting
- change in sense of taste, loss of taste
- loss of appetite, weight loss
- change in skin colour
- change in hair colour
- tingling or rash on palms of hands or soles of feet
- rash, dry skin, skin redness, scaly skin, itchy skin, blisters; skin infections, pus formation, skin ulcers
- headache
- constipation
- sore tongue, sore mouth, dry mouth, difficulty swallowing, cold sores
- cough
- upset stomach, stomach pain, wind, heart burn, indigestion
- pain in fingers, arms or legs
- weakness
- muscle pain, joint pain, back pain
- dizziness
- hair loss
- nose bleed
- increased tears, watery eyes
- tingling or numbness of hands or feet; pins and needles
- difficulty sleeping
- depression
- unusual urine colour, frequency or pain passing urine
- swelling, weight gain, enlargement of thyroid gland
- feeling overheated, increased sweating
- fast or irregular heart beat.
These are the more common side effects.
Tell your doctor immediately if you get any of the following side effects:
- shortness of breath, wheezing or trouble breathing, chest pain
- swelling of feet or legs, leg pain
- swollen face, eyelids, lip, tongue or voice box; swelling under the skin
- bleeding or bruising under the skin; coughing blood
- flu-like symptoms (chills, fever, sore throat, swollen glands)
- high blood pressure
- very bad stomach pain
- leaking or discharge near anus
- fits, seizures
- infection
- swelling, dark marks or blisters on any part of the body
- muscle pain, weakness or wasting
- decrease in amount of urine
- yellowing of skin and eyes (jaundice)
- numbness or tingling on one side of the body; weakness of face, arm or leg; trouble speaking, seeing or swallowing; headache, confusion, dizziness, loss of co-ordination or balance
- pain or numbness in the jaw, teeth or gums
- decreased blood sugar level, feeling hungry, shaky or anxious
- pain in the neck, shoulder or arm
- pressure in the chest
- lack of energy, confusion, sleepiness, decline in mental abilities, changes in consciousness (signs of brain toxicity due to high blood levels of ammonia).
The above side effects may be serious. You may need urgent medical attention.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some patients.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using SUTENT
Storage
Keep your capsules in the original container until it is time to take them.
Store SUTENT in a cool dry place.
Do not leave SUTENT or any other medicine in the car or on window sills.
Do not store SUTENT or any other medicine in the bathroom or near a sink. Heat and dampness can destroy some medicines.
Keep the capsules where children cannot reach them. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any capsules you have left.
Product description
What it looks like
SUTENT 12.5 mg capsules have a Swedish Orange (a brownish red colour) cap and body and are printed with "Pfizer" on the cap and "STN 12.5mg" on the body in white ink.
SUTENT 25 mg capsules have a caramel-coloured cap and Swedish Orange (a brownish red colour) body and are printed with "Pfizer" on the cap and "STN 25mg" on the body in white ink.
SUTENT 37.5 mg capsules have a yellow cap and body and are printed with "Pfizer" on the cap and "STN 37.5mg" on the body in black ink.
SUTENT 50 mg capsules have a caramel-coloured cap and body and are printed with "Pfizer" on the cap and "STN 50mg" on the body in white ink.
Each blister pack contains 28 capsules.
Ingredients
Sutent capsules contain sunitinib malate equivalent to 12.5 mg, 25 mg, 37.5 mg or 50 mg of sunitinib.
The capsules also contain:
- mannitol
- croscarmellose sodium
- povidone
- magnesium stearate
- gelatin
- titanium dioxide (E171)
- red iron oxide CI77491 (E172) (12.5 mg, 25 mg and 50 mg)
- yellow iron oxide CI77492 (E172) (25 mg, 37.5 mg and 50 mg)
- black iron oxide CI77499 (E172) (25 mg and 50 mg).
SUTENT capsules do not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Supplier
SUTENT is supplied in Australia by:
Pfizer Australia Pty Ltd
Sydney NSW
Toll Free Number: 1800 675 229
www.pfizermedicalinformation.com.au
Australian Registration Numbers
SUTENT 12.5 mg:
AUST R 149114 (blister pack)
SUTENT 25 mg:
AUST R 149115 (blister pack)
SUTENT 37.5 mg:
AUST R 156801 (blister pack)
SUTENT 50 mg:
AUST R 149116 (blister pack)
Date of preparation
This leaflet was prepared in February 2025.
© Pfizer Australia Pty Ltd
® = Registered trademark
Published by MIMS April 2025
 In the double-blind treatment phase of the GIST study, oral pain other than mucositis/stomatitis occurred in 12 patients (6%) on Sutent versus 3 (3%) on placebo. Hair colour changes occurred in 15 patients (7%) on Sutent versus 4 (4%) on placebo. Alopecia was observed in 10 patients (5%) on Sutent versus 2 (2%) on placebo.
In the double-blind treatment phase of the GIST study, oral pain other than mucositis/stomatitis occurred in 12 patients (6%) on Sutent versus 3 (3%) on placebo. Hair colour changes occurred in 15 patients (7%) on Sutent versus 4 (4%) on placebo. Alopecia was observed in 10 patients (5%) on Sutent versus 2 (2%) on placebo.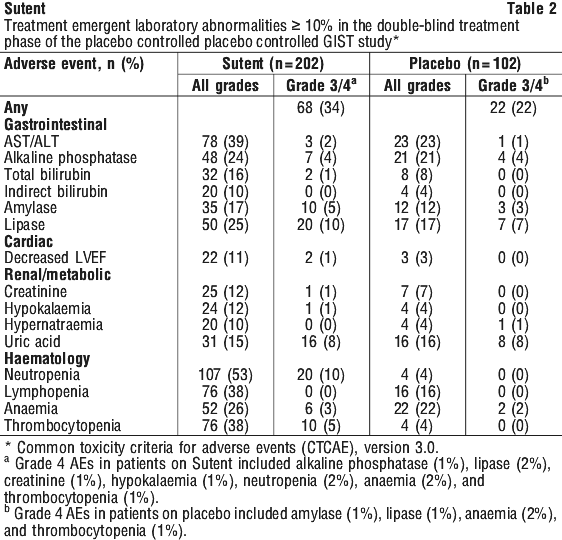 Grade 3 or 4 treatment emergent laboratory abnormalities were observed in 68 (34%) versus 22 (22%) patients on Sutent and placebo, respectively. Elevated liver function tests, pancreatic enzymes and creatinine were more common in patients treated with Sutent than placebo. Decreased LVEF and myelosuppression were also more common with Sutent treatment. Treatment emergent electrolyte disturbances of all types were more common in patients on Sutent than on placebo, including hyperkalaemia (6% vs. 4%), hypokalaemia (12% vs. 4%), hypernatraemia (10% vs. 4%), hyponatraemia (6% vs. 1%) and hypophosphataemia (9% vs. 0%). Three Sutent patients (1.5%) had grade 3 hypophosphataemia. Acquired hypothyroidism was noted in 8 patients (4%) on Sutent versus 1 (1%) on placebo.
Grade 3 or 4 treatment emergent laboratory abnormalities were observed in 68 (34%) versus 22 (22%) patients on Sutent and placebo, respectively. Elevated liver function tests, pancreatic enzymes and creatinine were more common in patients treated with Sutent than placebo. Decreased LVEF and myelosuppression were also more common with Sutent treatment. Treatment emergent electrolyte disturbances of all types were more common in patients on Sutent than on placebo, including hyperkalaemia (6% vs. 4%), hypokalaemia (12% vs. 4%), hypernatraemia (10% vs. 4%), hyponatraemia (6% vs. 1%) and hypophosphataemia (9% vs. 0%). Three Sutent patients (1.5%) had grade 3 hypophosphataemia. Acquired hypothyroidism was noted in 8 patients (4%) on Sutent versus 1 (1%) on placebo. Other significant adverse events occurring in cytokine refractory mRCC patients receiving Sutent included peripheral neuropathy (10%), appetite disturbance (9%), blistering of the skin (7%), periorbital oedema (7%) and increased lacrimation (6%).
Other significant adverse events occurring in cytokine refractory mRCC patients receiving Sutent included peripheral neuropathy (10%), appetite disturbance (9%), blistering of the skin (7%), periorbital oedema (7%) and increased lacrimation (6%).
 Table 6 provides common (≥ 10%) treatment emergent laboratory abnormalities.
Table 6 provides common (≥ 10%) treatment emergent laboratory abnormalities.

 Sunitinib treatment was associated with longer survival compared to IFN-α (see Figure 2). The median OS was 114.6 weeks for the sunitinib arm (95% CI: 100.1, 142.9) and 94.9 weeks for the IFN-α arm (95% CI: 77.7, 117.0) [HR: 0.821; 95% CI: 0.673, 1.001; p = 0.0510 by log-rank test, p = 0.013 by Wilcoxon test]. In the stratified analysis (LDH > versus ≤ 1.5 x ULN, ECOG performance status 0 versus ≥ 1, and absence or presence of prior nephrectomy), the HR was 0.818 (95% CI: 0.669, 0.999; p = 0.049 by log-rank test). The median OS for the IFN-α arm includes 25 subjects who discontinued IFN-α treatment because of disease progression and crossed over to treatment with sunitinib. Following discontinuation from the study, 213 subjects on the IFN-α arm received poststudy cancer treatment, including 32% who received sunitinib; 182 subjects on the sunitinib arm received poststudy cancer treatment, including 11% who received sunitinib. In post-hoc analyses censoring subjects who crossed over from IFN-α treatment to sunitinib treatment, median OS at the time of crossover was 114.6 versus 86.7 weeks (unstratified hazard ratio: 0.808; p = 0.0361 by log-rank test; p = 0.0081 by Wilcoxon test). When excluding subjects who received poststudy anticancer therapy, median OS was 121.9 versus 61.3 weeks on sunitinib versus IFN-α respectively (HR: 0.647; 95% CI: 0.482, 0.867; p = 0.0033 by log-rank test; p = 0.0013 by Wilcoxon test).
Sunitinib treatment was associated with longer survival compared to IFN-α (see Figure 2). The median OS was 114.6 weeks for the sunitinib arm (95% CI: 100.1, 142.9) and 94.9 weeks for the IFN-α arm (95% CI: 77.7, 117.0) [HR: 0.821; 95% CI: 0.673, 1.001; p = 0.0510 by log-rank test, p = 0.013 by Wilcoxon test]. In the stratified analysis (LDH > versus ≤ 1.5 x ULN, ECOG performance status 0 versus ≥ 1, and absence or presence of prior nephrectomy), the HR was 0.818 (95% CI: 0.669, 0.999; p = 0.049 by log-rank test). The median OS for the IFN-α arm includes 25 subjects who discontinued IFN-α treatment because of disease progression and crossed over to treatment with sunitinib. Following discontinuation from the study, 213 subjects on the IFN-α arm received poststudy cancer treatment, including 32% who received sunitinib; 182 subjects on the sunitinib arm received poststudy cancer treatment, including 11% who received sunitinib. In post-hoc analyses censoring subjects who crossed over from IFN-α treatment to sunitinib treatment, median OS at the time of crossover was 114.6 versus 86.7 weeks (unstratified hazard ratio: 0.808; p = 0.0361 by log-rank test; p = 0.0081 by Wilcoxon test). When excluding subjects who received poststudy anticancer therapy, median OS was 121.9 versus 61.3 weeks on sunitinib versus IFN-α respectively (HR: 0.647; 95% CI: 0.482, 0.867; p = 0.0033 by log-rank test; p = 0.0013 by Wilcoxon test).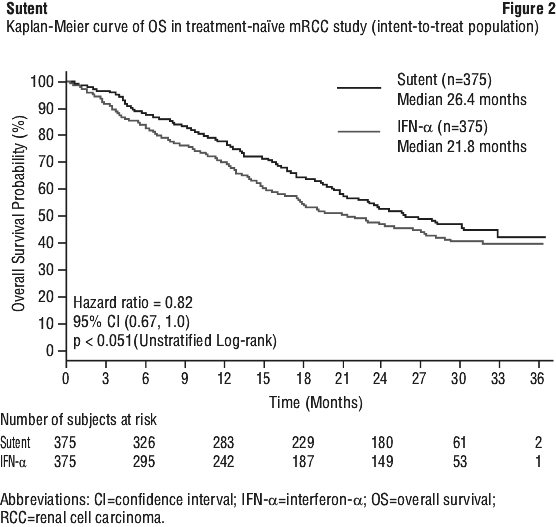 Patient-reported outcomes (PRO) were measured using the Functional Assessment of Cancer Therapy-Advanced Kidney Cancer Symptom Index (FKSI) and the Functional Assessment of Cancer Therapy-General (FACT-G). PRO endpoints include the FKSI score, its Disease Related Symptoms subscale (FKSI-DRS) score, the FACT-G total score and its four subscale scores (Physical Well-Being [PWB], Social/Family Well-Being [SWB], Emotional Well-Being [EWB] and Functional Well Being [FWB]. The FKSI-DRS was pre-specified as the primary PRO endpoint and used to assess patient-reported kidney cancer related symptoms (lack of energy/fatigue, pain/bone pain, weight loss, shortness of breath, cough, fever, and haematuria) in 719 subjects. Subjects treated with sunitinib reported statistically significant better FKSI-DRS index scores (p ≤ 0.0071), FKSI scores (p ≤ 0.0133), FACT-G total scores (p ≤ 0.0244), PWB (p ≤ 0.0208), and FWB (p ≤ 0.0044) than subjects treated with IFN-α at all postbaseline assessment time points up to 20 cycles of treatment. For PWB, SWB, and EWB, the statistical significance level increased above the 0.05 level after cycle 13, cycle 15 day 1, and cycle 10 respectively. Compared to the pre-established minimum clinically important differences for these endpoints, the between treatment differences for kidney cancer related symptoms (FKSI at all postbaseline timepoints and FKSI DRS after cycle 3, day 1) and overall quality of life (FACT-G) at all postbaseline time points were considered clinically meaningful.
Patient-reported outcomes (PRO) were measured using the Functional Assessment of Cancer Therapy-Advanced Kidney Cancer Symptom Index (FKSI) and the Functional Assessment of Cancer Therapy-General (FACT-G). PRO endpoints include the FKSI score, its Disease Related Symptoms subscale (FKSI-DRS) score, the FACT-G total score and its four subscale scores (Physical Well-Being [PWB], Social/Family Well-Being [SWB], Emotional Well-Being [EWB] and Functional Well Being [FWB]. The FKSI-DRS was pre-specified as the primary PRO endpoint and used to assess patient-reported kidney cancer related symptoms (lack of energy/fatigue, pain/bone pain, weight loss, shortness of breath, cough, fever, and haematuria) in 719 subjects. Subjects treated with sunitinib reported statistically significant better FKSI-DRS index scores (p ≤ 0.0071), FKSI scores (p ≤ 0.0133), FACT-G total scores (p ≤ 0.0244), PWB (p ≤ 0.0208), and FWB (p ≤ 0.0044) than subjects treated with IFN-α at all postbaseline assessment time points up to 20 cycles of treatment. For PWB, SWB, and EWB, the statistical significance level increased above the 0.05 level after cycle 13, cycle 15 day 1, and cycle 10 respectively. Compared to the pre-established minimum clinically important differences for these endpoints, the between treatment differences for kidney cancer related symptoms (FKSI at all postbaseline timepoints and FKSI DRS after cycle 3, day 1) and overall quality of life (FACT-G) at all postbaseline time points were considered clinically meaningful. The primary endpoint for both studies was ORR. The core imaging laboratory reported 38 partial responses (PRs) in the pivotal study resulting in an ORR of 35.8% (95% CI: 26.8, 45.7). Consistent results were observed in the supportive study where an ORR of 25.4% was demonstrated. The majority of objective disease responses were observed during cycles 2 to 4; responses were observed as late as cycle 11. Duration of tumour response (DR) data from the pivotal study is premature as only a relatively small number of patients responding to treatment had experienced disease progression (median DR not yet reached (95% CI: 42.0 weeks*) using core laboratory assessment). The median DR in the supportive study, based on investigator assessment, was 54 weeks (95% CI: 34.3, 70.1). These results indicate that disease responses induced by Sutent in patients with cytokine refractory RCC were durable.
The primary endpoint for both studies was ORR. The core imaging laboratory reported 38 partial responses (PRs) in the pivotal study resulting in an ORR of 35.8% (95% CI: 26.8, 45.7). Consistent results were observed in the supportive study where an ORR of 25.4% was demonstrated. The majority of objective disease responses were observed during cycles 2 to 4; responses were observed as late as cycle 11. Duration of tumour response (DR) data from the pivotal study is premature as only a relatively small number of patients responding to treatment had experienced disease progression (median DR not yet reached (95% CI: 42.0 weeks*) using core laboratory assessment). The median DR in the supportive study, based on investigator assessment, was 54 weeks (95% CI: 34.3, 70.1). These results indicate that disease responses induced by Sutent in patients with cytokine refractory RCC were durable. At the time of a pre-specified interim analysis, a statistically significant prolongation in the primary endpoint, TTP, was observed between the treatment arms and was considered clinically significant (Figure 3). The median TTP by core imaging laboratory assessment was 27.3 vs. 6.4 weeks for the Sutent and placebo arms, respectively (HR: 0.329, 95% CI: 0.222, 0.466, p-value < 0.00001). The risk of experiencing progression was 3 times higher for patients in the placebo arm compared to the Sutent arm (representing a 72% reduction in the risk of developing progressive disease for patients receiving Sutent). Median TTP for the group of patients treated with Sutent was more than 4 times longer than that for patients receiving placebo. Results of the dose escalating study with median TTP of 34.0 weeks by investigator assessment are consistent with the results of the phase 3 study.
At the time of a pre-specified interim analysis, a statistically significant prolongation in the primary endpoint, TTP, was observed between the treatment arms and was considered clinically significant (Figure 3). The median TTP by core imaging laboratory assessment was 27.3 vs. 6.4 weeks for the Sutent and placebo arms, respectively (HR: 0.329, 95% CI: 0.222, 0.466, p-value < 0.00001). The risk of experiencing progression was 3 times higher for patients in the placebo arm compared to the Sutent arm (representing a 72% reduction in the risk of developing progressive disease for patients receiving Sutent). Median TTP for the group of patients treated with Sutent was more than 4 times longer than that for patients receiving placebo. Results of the dose escalating study with median TTP of 34.0 weeks by investigator assessment are consistent with the results of the phase 3 study.
 The final ITT population enrolled in the double-blind treatment phase of the study included 243 patients randomised to the sunitinib arm and 118 subjects randomised to the placebo arm. After the primary endpoint was met at the interim analysis, the study was unblinded, and patients on the placebo arm were offered open-label sunitinib treatment.
The final ITT population enrolled in the double-blind treatment phase of the study included 243 patients randomised to the sunitinib arm and 118 subjects randomised to the placebo arm. After the primary endpoint was met at the interim analysis, the study was unblinded, and patients on the placebo arm were offered open-label sunitinib treatment. Of those patients randomised to the sunitinib arm, 62.7% survived longer than 1 year, 35.5% survived longer than 2 years, and 22.3% survived longer than 3 years.
Of those patients randomised to the sunitinib arm, 62.7% survived longer than 1 year, 35.5% survived longer than 2 years, and 22.3% survived longer than 3 years.

 OS data were not mature at the time of the analysis. There were 21 deaths in the Sutent arm and 30 deaths in the placebo arm. Patients in the placebo arm were able to receive Sutent after disease progression, possibly confounding the survival analysis. A statistically significant difference in ORR favouring Sutent over placebo was observed.
OS data were not mature at the time of the analysis. There were 21 deaths in the Sutent arm and 30 deaths in the placebo arm. Patients in the placebo arm were able to receive Sutent after disease progression, possibly confounding the survival analysis. A statistically significant difference in ORR favouring Sutent over placebo was observed. Chemical name: (Z)-N-[2-(Diethylamino)ethyl]-5-[(5-fluoro-2-oxo-1,2-dihydro-3H-indol-3-ylidene) methyl]-2, 4-dimethyl-1H-pyrrole-3-carboxamide (S)-2-hydroxysuccinate.
Chemical name: (Z)-N-[2-(Diethylamino)ethyl]-5-[(5-fluoro-2-oxo-1,2-dihydro-3H-indol-3-ylidene) methyl]-2, 4-dimethyl-1H-pyrrole-3-carboxamide (S)-2-hydroxysuccinate.