What is in this leaflet
This leaflet answers some common questions about TALMINEX capsule.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking TALMINEX against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What TALMINEX is used for
TALMINEX is an antiviral medicine containing the active ingredient oseltamivir.
TALMINEX is used for the treatment and prevention of influenza (an infection caused by the influenza virus). It has no effect on the common cold or other respiratory virus infections.
TALMINEX belongs to a group of medicines called neuraminidase inhibitors. These medicines attack the influenza virus and prevent it from spreading inside your body.
TALMINEX is absorbed to the key sites of influenza infection and treats the cause. Taking TALMINEX means you feel better faster.
You will also be less likely to develop complications of influenza, such as bronchitis, pneumonia and sinusitis.
Typical symptoms of influenza include fever, headache, muscle aches, sore throat, cough and generally feeling unwell.
Ask your doctor if you have any questions about why TALMINEX has been prescribed for you.
TALMINEX is not addictive.
This medicine is available only with a doctor's prescription.
Ask your doctor about having the influenza vaccination. Vaccination every year is the best way to prevent influenza.
Before you take TALMINEX
When you must not take it
Do not TALMINEX if:
- you have had an allergic reaction to oseltamivir or any ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- the package is torn or shows signs of tampering
- the expiry date (EXP) printed on the pack has passed.
If you take this medicine after the expiry date has passed, it may not work as well.
If you are not sure if you should be taking TALMINEX, talk to your doctor.
Use in the elderly
Although there is limited experience with use of TALMINEX in patients 65 years and older, the dose recommended for use in elderly patients is the same as that recommended for adults.
Use in children
Do not give TALMINEX to children under 1 year of age for the prevention of influenza. Safety and effectiveness of oseltamivir in children under 1 year of age have not been established when used for the prevention of influenza.
Before you start to take it
Tell your doctor if:
- you are pregnant or plan to become pregnant
It is not known whether oseltamivir is harmful to an unborn baby when taken by a pregnant woman. If there is a need to take oseltamivir when you are pregnant your doctor will discuss the risks and benefits to you and the unborn baby
- you are breast-feeding or plan to breast-feed
Oseltamivir may pass into breast milk. Your doctor will discuss the risks and benefits of using oseltamivir if you are breast-feeding.
- you have any other health problems, especially the following:
kidney failure, kidney impairment or kidney disease.
if you have a weakened immune system, caused by a medical condition or medication you are taking.
- you are allergic to any other medicines, foods, dyes or preservatives
If you have not told your doctor about any of the above, tell them before you start taking TALMINEX.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought from a pharmacy, supermarket or health food shop. These medicines may be affected by TALMINEX or may affect how well it works. You may need to use different amounts of your medicine, or you may need to take different medicines. Your doctor will advise you.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking TALMINEX including:
- immunosuppressants, medicines used to suppress the immune system
- probenecid, a medicine used to treat gout
It is safe to take aspirin, paracetamol and cough medicines with TALMINEX. However, medical advice should be sought before giving aspirin to children with viral illness.
How to take TALMINEX capsules
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
Start taking TALMINEX as soon as possible within the first two days of the onset of the first symptoms of influenza or exposure to someone with influenza. The earlier you start treatment with TALMINEX, the shorter the duration of your influenza.
How much to take
Take TALMINEX exactly as your doctor has prescribed.
If you have kidney disease your doctor may prescribe you a lower dose of TALMINEX.
TREATMENT OF INFLUENZA
Adults and adolescents
The recommended oral dose of TALMINEX for adults and adolescents is 75 mg twice a day for 5 days.
Children under 1 year of age
Oseltamivir 6 mg/mL powder for oral suspension (available in other brands) is the preferred product (see separate oseltamivir 6 mg/mL powder for oral suspension Consumer Medicine Information).
Children 1 year of age or older
Give TALMINEX as directed by your child's doctor or pharmacist.
The usual dose of TALMINEX is one dose taken TWICE a day for 5 days. The dose may vary depending on your child's weight.
PREVENTION OF INFLUENZA
For prevention of influenza, TALMINEX capsules are taken once a day at the recommended dose while protection is required. Safety and effectiveness have been shown in patients taking TALMINEX for up to 6 weeks.
Adults and adolescents
The recommended oral prevention dose of TALMINEX for adults and adolescents 13 years and older is 75 mg once a day for 10 days.
Children 1 year of age or older
Give TALMINEX as directed by your child's doctor or pharmacist.
The usual dose of TALMINEX is one dose taken ONCE a day for 10 days. The dose may vary depending on your child's weight
Do not give TALMINEX to children under 1 year of age for the prevention of influenza. Safety and effectiveness in children under 1 year of age have not been established.
How to take it
Swallow capsules whole with a glass of water with or without food.
It does not matter whether you take TALMINEX with food or not. However, if TALMINEX upsets your stomach, it is better to take TALMINEX with food.
Do not break or chew the capsules before swallowing.
If you cannot swallow the capsule whole:
For adults, adolescents or children 1 year of age or older who are unable to swallow capsules please follow these instructions to ensure proper dosing:
- hold the required dosage capsule over a small bowl, carefully pull the capsule open and pour the powder into the bowl
- add a suitable, small amount (1 teaspoon maximum) of sweetened food product such as regular or sugar-free chocolate syrup, honey (only for children two years or older), light brown or table sugar dissolved in water, dessert toppings, sweetened condensed milk, apple sauce or yogurt to mask the bitter taste of the medicine
- stir the mixture well and give the entire contents of the bowl to the patient. The mixture must be swallowed immediately after its preparation. If there is some mixture left inside the bowl, rinse the bowl with a small amount of water and have the patient drink this remaining mixture.
For children 1 year of age or older requiring doses different to that available in capsule form, please follow these instructions to ensure proper dosing:
- hold one TALMINEX 75 mg capsule over a small bowl, carefully pull the capsule open and pour the powder into the bowl
- add 5 mL water to the powder using a syringe with markings (called a "graduated syringe") to show how much fluid has been drawn up. Stir for about two minutes
- draw up into the syringe the correct amount of mixture from the bowl based on the recommended dose required (see below), which is body weight dependent.
It is not necessary to draw up any undissolved white powder.
- Recommended dose 30mg
Amount of TALMINEX mixture for one dose - 2mL - Recommended dose 45mg
Amount of TALMINEX mixture for one dose - 3mL - Recommended dose 60mg
Amount of TALMINEX mixture for one dose - 4mL
Push down on the plunger of the syringe, to empty its entire contents into a second bowl.
Discard any unused mixture.
- Immediately after administration, take the dispenser apart and rinse both parts of the dispenser under running tap water. Air dry prior to next use.
- in the second bowl, add a suitable, small amount (1 teaspoon maximum) of sweetened food product to the mixture to mask the bitter taste of the medicine
- stir this mixture well and give the entire contents of the second bowl to the patient. This mixture must be swallowed immediately after its preparation. If there is some mixture left inside the bowl, rinse the bowl with a small amount of water and have the patient drink this remaining mixture.
The appropriate dose must be mixed by the caregiver with an equal quantity of sweetened food product such as regular or sugar-free chocolate syrup, light brown or table sugar dissolved in water, dessert toppings, sweetened condensed milk, apple sauce or yoghurt to mask the bitter taste of the medicine.
Patients who are unable to swallow capsules may also receive oseltamivir 6 mg/mL powder for oral suspension (available in other brands).
When to take it
Treatment with TALMINEX should be started as soon as possible, but no later than 48 hours after the first symptoms of influenza. For influenza treatment, TALMINEX should be taken in the morning and in the evening. For influenza prevention, TALMINEX should be taken once a day.
If you have kidney problems, your doctor may tell you take TALMINEX less often.
Taking your medicine at the same time each day will help you remember when to take the capsules.
How long to take it
Continue taking TALMINEX until your doctor tells you to stop or your course of treatment is complete.
If you have a weakened immune system, your doctor may tell you to take a longer course.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember and then go back to taking it as you would normally.
If you are not sure what to do, ask your doctor or pharmacist. If you have trouble remembering your dose, ask your pharmacist for some hints.
In case of an overdose
If you think you or anyone else may have taken too much TALMINEX immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26) or go to Accident and Emergency at your nearest hospital. Do this even if there are no signs of discomfort or poisoning. Keep telephone numbers for these places handy. If you are not sure what to do, contact your doctor or pharmacist.
The following are some symptoms of overdose which may or may not occur:
- nausea (feeling like vomiting)
- vomiting
While you are taking TALMINEX
Things you must do:
Tell your doctor if you have kidney failure or impairment or any other problems with your kidneys.
Tell all doctors, dentists and pharmacists who are treating you that you are taking TALMINEX.
Tell your doctor if you become pregnant while taking TALMINEX.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Tell your doctor if you feel your symptoms have worsened after starting TALMINEX.
Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Things you must not do
Do not stop taking TALMINEX or change the dose without first checking with your doctor.
Do not let yourself run out of medicine over the weekend or on holidays.
Do not give TALMINEX to anyone else even if they have the same condition as you.
Do not use TALMINEX to treat other complaints unless your doctor says to.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how TALMINEX affects you. However, TALMINEX is not expected to affect your ability to drive a car or operate machinery.
Side Effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TALMINEX.
TALMINEX helps most people with influenza but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you.
- nausea (feeling like vomiting)
- vomiting
- dizziness/spinning sensation (vertigo)
- headache
- stomach ache, indigestion
- diarrhoea
- cough
- bronchitis
- asthma (breathlessness, wheezing, a cough sometimes brought on by exercise and a feeling of tightness in the chest)
- sinusitis (stuffy nose and/or feeling of tension or fullness in the nose, cheeks and behind the eyes, sometimes with a throbbing ache
- runny nose or nose bleeds
- ear problems or ear infection
- conjunctivitis (discharge from the eyes with itching and crusty eyelids)
- visual disturbances
- insomnia (difficulty sleeping)
- fatigue
- aches and pains
- mild skin rash
These are the more common side effects of TALMINEX. Mostly these are mild.
Taking TALMINEX with food may reduce the potential for some or all of these side effects.
Tell your doctor immediately, or go to the Accident and Emergency department if you notice any of the following:
- sudden signs of allergy such as rash, itching or hives on the skin, swelling of the face, lips, tongue or other parts of the body, shortness of breath, wheezing or trouble breathing
- yellowing of the skin and/or eyes, itching and dark coloured urine
- chest infection with fever, chills, shortness of breath, cough, phlegm and occasional blood
- convulsions, confusion, drowsiness, abnormal behaviour, delusions, hallucinations, agitation, anxiety and nightmares. These symptoms may also occur in influenza patients not treated with TALMINEX.
Patients, especially children and adolescents, should be closely monitored and their healthcare professional should be contacted immediately if the patient shows any signs of unusual behaviour.
- Diarrhoea with blood, along with fever and severe stomach pain
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor or pharmacist if you don't understand anything on this list.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After taking TALMINEX
Storage
Keep your capsules in the blister pack until it is time to take them. If you take the capsules out of the blister pack they may not keep well.
Keep your capsules in a cool dry place where the temperature stays below 25ºC.
Do not store it or any other medicine in a bathroom, or near a sink.
Do not leave it in the car or on window sills. Heat and damp can destroy some medicines.
Keep TALMINEX where young children cannot reach them. A locked cupboard at least one-and-a half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking TALMINEX or the capsules have passed their expiry date, ask your pharmacist what to do with any capsules that are left over.
Product description
Availability
TALMINEX is available in the following strengths: 30 mg, 45 mg and 75 mg.
TALMINEX comes in blister packs containing 10 capsules.
What it looks like
TALMINEX 30 mg capsules have a light yellow opaque colour body with black colour band, imprinted with “M” and light yellow opaque colour cap imprinted with “30 mg” (AUST R 312888)*.
TALMINEX 45 mg capsules have a grey opaque colour body with black colour band, imprinted with “M” and a grey opaque colour cap imprinted with “45 mg” (AUST R 312889)*.
TALMINEX 75 mg capsules have a grey opaque colour body with black colour band, imprinted with “M” and a light yellow opaque colour cap imprinted with “75 mg” (AUST R 312887).
*TALMINEX 30 mg and 45 mg capsules are not marketed for use in Australia.
Ingredients
Active ingredient - oseltamivir
- 30 mg capsules contain 30 mg oseltamivir (present as 39.4 mg oseltamivir phosphate).
- 45 mg capsules contain 45 mg oseltamivir (present as 59.1 mg oseltamivir phosphate).
- 75 mg capsules contain 75 mg oseltamivir (present as 98.5 mg oseltamivir phosphate)
Inactive ingredients
- Capsule contents:
- pregelatinised maize starch
- povidone
- croscarmellose sodium
- purified talc
- sodium stearyl fumarate - Capsule shell:
- gelatin
- titanium dioxide (171)
- black iron oxide (172)
- red iron oxide (172)
- yellow iron oxide (172)
- black ink (TekPrint SW-9008)
TALMINEX contain sulfites.
Sponsor
Accelagen Pty Ltd
Suite 2.02
785 Toorak Road
Hawthorn East
Victoria 3123 Australia
Distributor
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30 – 34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in November 2023.
TALMINEX® is a Viatris company trade mark
Published by MIMS July 2024
 Children ≥ 1 year old who are able to swallow capsules may receive treatment with 30 mg, 45 mg or 75 mg capsules (one 30 mg capsule plus one 45 mg capsule may be used in place of a 75 mg capsule) twice daily.
Children ≥ 1 year old who are able to swallow capsules may receive treatment with 30 mg, 45 mg or 75 mg capsules (one 30 mg capsule plus one 45 mg capsule may be used in place of a 75 mg capsule) twice daily.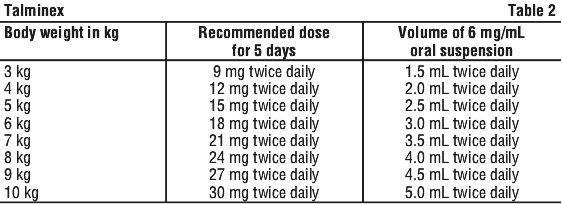 * This table is not intended to contain all possible weights for this population. For all patients under the age of 1 year of age, 3 mg/kg should be used to determine dose regardless of the weight of the patient.
* This table is not intended to contain all possible weights for this population. For all patients under the age of 1 year of age, 3 mg/kg should be used to determine dose regardless of the weight of the patient. Children ≥ 1 year old who are able to swallow capsules may receive treatment with 30 mg, 45 mg or 75 mg capsules. A 75 mg dose may be achieved with a 75 mg capsule once daily or one 30 mg capsule plus one 45 mg capsule once daily.
Children ≥ 1 year old who are able to swallow capsules may receive treatment with 30 mg, 45 mg or 75 mg capsules. A 75 mg dose may be achieved with a 75 mg capsule once daily or one 30 mg capsule plus one 45 mg capsule once daily. 4. In the second bowl, add a suitable, small amount (1 teaspoon maximum) of sweetened food product such as regular or sugar-free chocolate syrup, honey (only for children two years or older), light brown or table sugar dissolved in water, dessert toppings, sweetened condensed milk, apple sauce or yogurt to the mixture to mask the bitter taste of the medication.
4. In the second bowl, add a suitable, small amount (1 teaspoon maximum) of sweetened food product such as regular or sugar-free chocolate syrup, honey (only for children two years or older), light brown or table sugar dissolved in water, dessert toppings, sweetened condensed milk, apple sauce or yogurt to the mixture to mask the bitter taste of the medication.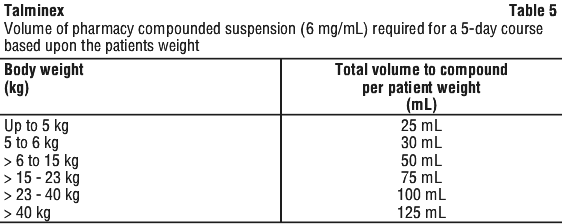 Second, determine the number of capsules and the amount of vehicle (water containing 0.05% w/v sodium benzoate added as a preservative) that is needed to prepare the total volume (calculated from the table above: 25 mL, 30 mL, 50 mL, 75 mL, 100 mL or 125 mL) of compounded Talminex 6 mg/mL suspension as shown in Table 6.
Second, determine the number of capsules and the amount of vehicle (water containing 0.05% w/v sodium benzoate added as a preservative) that is needed to prepare the total volume (calculated from the table above: 25 mL, 30 mL, 50 mL, 75 mL, 100 mL or 125 mL) of compounded Talminex 6 mg/mL suspension as shown in Table 6.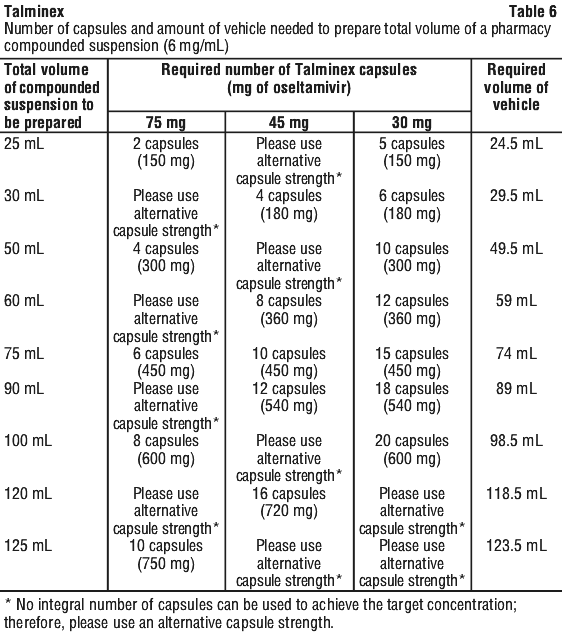 Third, follow the procedure below for compounding the suspension (6 mg/mL) from Talminex capsules:
Third, follow the procedure below for compounding the suspension (6 mg/mL) from Talminex capsules: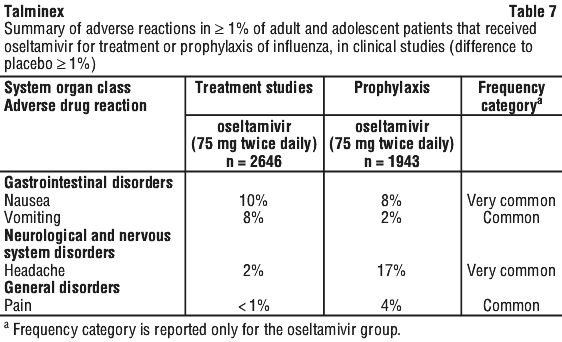
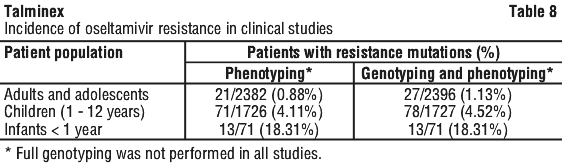
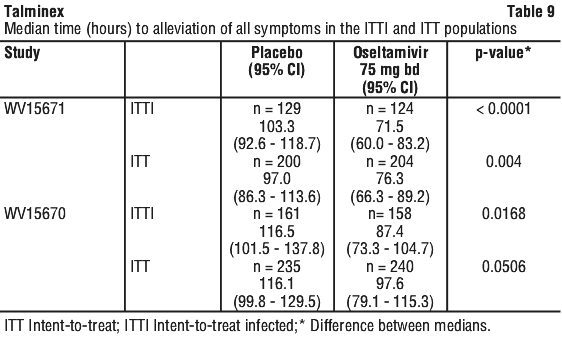
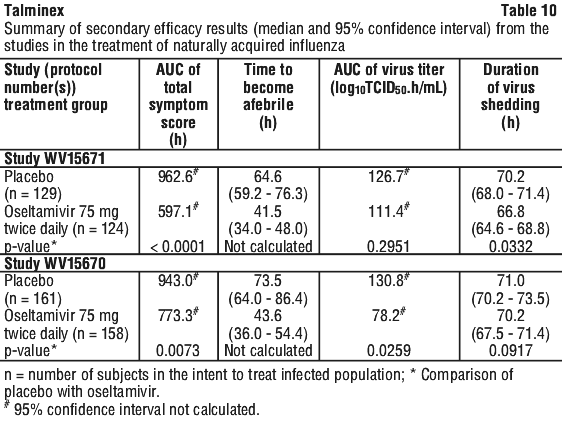
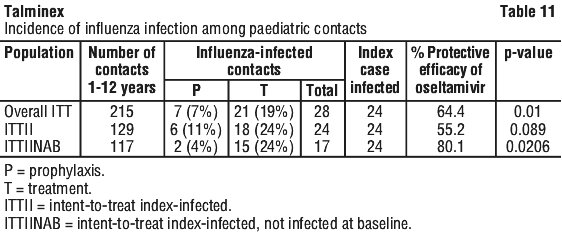
 Chemical name: (3R,4R,5S)-4-acetylamino-5-amino-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylic acid, ethyl ester, phosphate (1:1).
Chemical name: (3R,4R,5S)-4-acetylamino-5-amino-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylic acid, ethyl ester, phosphate (1:1).