1 Name of Medicine
Atezolizumab.
2 Qualitative and Quantitative Composition
Tecentriq 1875 mg/15 mL solution for subcutaneous (SC) injection.
Each vial of 15 mL contains 1875 mg of atezolizumab at a concentration of 125 mg/mL.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Solution for subcutaneous injection.
Tecentriq SC is supplied as a sterile, ready to use, single-dose vial containing 15 mL preservative-free, colourless to slightly yellow solution, at a concentration of 125 mg/mL.
Tecentriq SC formulation is not intended for intravenous administration. For information about the intravenous dosage form of Tecentriq please see the separate intravenous Tecentriq Product Information.4.1 Therapeutic Indications
Early-stage non-small cell lung cancer.
Tecentriq SC as monotherapy is indicated as adjuvant treatment following complete resection and no progression after platinum-based adjuvant chemotherapy for adult patients with stage II to IIIA (as per 7th edition of the UICC/AJCC staging system) NSCLC whose tumours have PD-L1 expression on ≥ 50% of tumour cells.
Metastatic non-small cell lung cancer.
Tecentriq SC, in combination with bevacizumab, paclitaxel and carboplatin, is indicated for the first-line treatment of adult patients with metastatic non-squamous non-small cell lung cancer (NSCLC). In patients with EGFR mutant or ALK-positive NSCLC, Tecentriq SC, in combination with bevacizumab, paclitaxel and carboplatin, is indicated only after failure of appropriate targeted therapies.
Tecentriq SC, in combination with nanoparticle albumin-bound paclitaxel (nab-paclitaxel) and carboplatin, is indicated for first-line treatment of patients with metastatic non-squamous NSCLC who do not have tumour EGFR or ALK genomic aberrations.
Tecentriq SC as monotherapy is indicated for the treatment of adult patients with locally advanced or metastatic NSCLC after prior chemotherapy. Patients with EGFR mutant or ALK-positive NSCLC should also have received targeted therapies before receiving Tecentriq SC.
Small cell lung cancer.
Tecentriq SC, in combination with carboplatin and etoposide, is indicated for the first-line treatment of patients with extensive-stage small cell lung cancer (ES-SCLC).
Urothelial carcinoma.
Tecentriq SC is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are considered cisplatin ineligible and whose tumours express PD-L1 (PD-L1 stained tumour-infiltrating immune cells [IC] covering ≥ 5% of the tumour area), as determined by a validated test.
This indication is approved based on overall response rate and duration of response in a single-arm study. Improvements in overall survival, progression-free survival, or health-related quality of life have not been established.
Hepatocellular carcinoma.
Tecentriq SC, in combination with bevacizumab, is indicated for the treatment of patients with unresectable or metastatic hepatocellular carcinoma (HCC) who have not received prior systemic therapy.4.2 Dose and Method of Administration
General.
Tecentriq SC must be initiated and supervised by physicians experienced in the treatment of cancer.
In order to improve the traceability of biological medicinal products, the trade name and the batch number of the administered product should be clearly recorded in the patient medical record.
It is important to check the product labels to ensure that the correct formulation (intravenous Tecentriq or Tecentriq SC) is being administered to the patient as prescribed.
Patients currently receiving intravenous Tecentriq.
Patients currently receiving intravenous Tecentriq can switch to Tecentriq SC (or vice versa).
Tecentriq SC.
Tecentriq SC formulation is not intended for intravenous administration.
Tecentriq SC must be administered as a subcutaneous injection into the thigh only (see Section 4.2, Instructions for administration).
Dose.
Tecentriq SC monotherapy. Patient selection for urothelial carcinoma.
Select cisplatin-ineligible patients with previously untreated locally advanced or metastatic urothelial carcinoma for treatment with Tecentriq SC based on the PD-L1 expression on tumour infiltrating immune cells confirmed by a validated test (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
See Table 1.
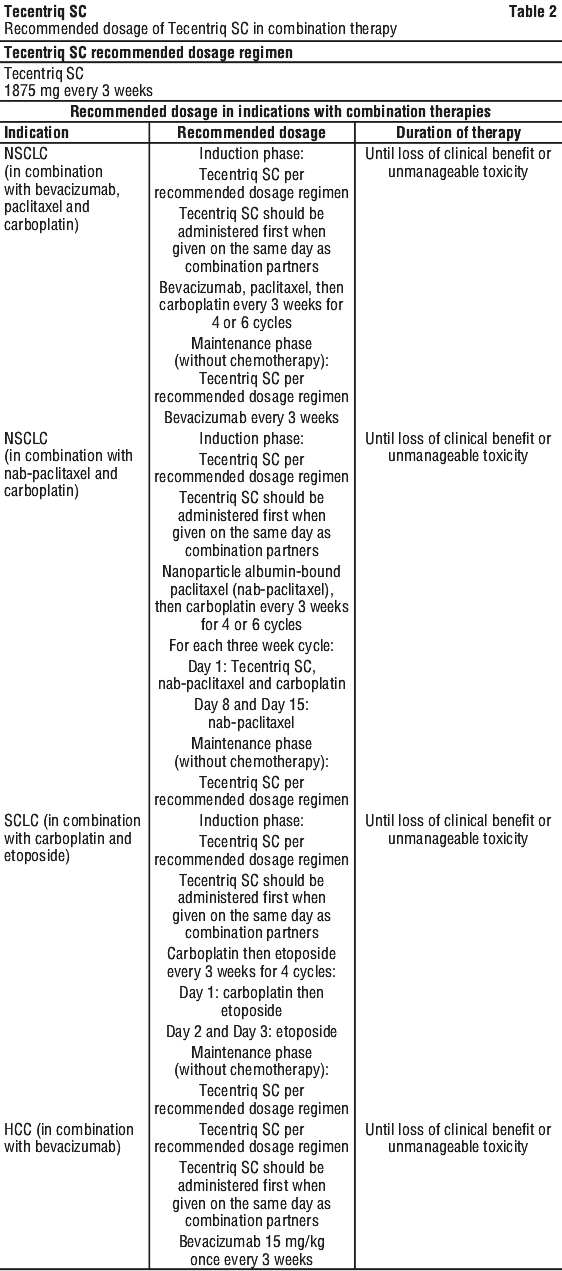
 Tecentriq SC in combination therapy. Please also refer to the Product Information for the combination products. See Table 2.
Tecentriq SC in combination therapy. Please also refer to the Product Information for the combination products. See Table 2.
Delayed or missed doses.
If a planned dose of Tecentriq SC is missed, it should be administered as soon as possible. The schedule of administration must be adjusted to maintain the appropriate interval between doses.
Dose modifications.
Dose reductions of Tecentriq SC are not recommended. See Table 3.
Dose delay or discontinuation.
Also see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects).
 Patients treated with Tecentriq SC must be given the Patient Card and be informed about the risks of atezolizumab.
Patients treated with Tecentriq SC must be given the Patient Card and be informed about the risks of atezolizumab.
Special dosage instructions.
Paediatric use.
The safety and efficacy of Tecentriq SC in children and adolescents below 18 years of age have not been established. Available safety data are described in Section 4.4.
Use in the elderly.
Based on a population pharmacokinetic analysis, no dose adjustment of Tecentriq SC is required in patients ≥ 65 years of age (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Use in Asian patients.
Due to increased haematologic toxicities observed in Asian patients in study GO29436 (IMpower150), it is recommended that the starting dose of paclitaxel should be 175 mg/m2 every three weeks.
Renal impairment.
Based on a population pharmacokinetic analysis, no dose adjustment is required in patients with mild or moderate renal impairment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties). Data from patients with severe renal impairment are too limited to draw conclusions on this population.
Hepatic impairment.
Based on a population pharmacokinetic analysis, no dose adjustment is required for patients with mild or moderate hepatic impairment. Atezolizumab has not been studied in patients with severe hepatic impairment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Eastern cooperative oncology group (ECOG) performance status ≥ 2.
Patients with ECOG performance status ≥ 2 were excluded from the clinical trials in NSCLC, ES-SCLC and HCC (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties).
Instructions for administration.
Tecentriq SC. Tecentriq SC is a ready-to-use solution for subcutaneous injection only and should not be diluted or mixed with other drugs.
Tecentriq SC should be inspected visually to ensure there is no particulate matter or discolouration prior to administration.
Tecentriq SC is for single use only and should be prepared by a healthcare professional.
Preparation of the syringe.
Tecentriq SC does not contain any antimicrobial preservative. If the dose is not administered immediately, see "Storage of the syringe" below.
Prior to use, remove the vial from the refrigerator and allow the solution to come to room temperature.
Withdraw the entire contents of Tecentriq SC solution from the vial with a syringe and transfer needle (18 G recommended).
Remove the transfer needle and attach a SC infusion set (e.g. winged/ butterfly) containing a 23-25 G stainless steel needle for injection. Use a SC infusion set with residual hold-up volume not exceeding 0.5 mL for administration.
Prime the SC infusion line with the drug product solution to eliminate the air in the infusion line and stop before the fluid reaches the needle.
Ensure the syringe contains exactly 15 mL of drug product solution after priming and expelling any excess volume from the syringe.
Administer immediately to avoid needle clogging. Do not store the prepared syringe that has been attached to the already-primed infusion set.
Administer Tecentriq SC solution subcutaneously in the thigh over approximately 7 minutes. Do not administer the remaining residual hold-up volume in the tubing to the patient.
The injection site should be alternated between the left and right thigh only. New injections should be given at least 2.5 cm from the previous site on healthy skin and never into areas where the skin is red, bruised, tender, or hard. During the treatment course with Tecentriq SC, other medications for subcutaneous administration should preferably be injected at different sites.
Storage of the syringe.
If the dose is not used immediately, use aseptic technique to withdraw the entire contents of Tecentriq SC solution from the vial into the syringe to account for the dose volume (15 mL) plus the priming volume for the SC infusion set. Replace the transfer needle with a syringe closing cap. Do not attach a SC infusion set for storage (see Section 6.3 Shelf Life).4.3 Contraindications
Tecentriq SC is contraindicated in patients with a known hypersensitivity to atezolizumab or any of the excipients.
4.4 Special Warnings and Precautions for Use
Assessment of PD-L1 status.
When assessing the PD-L1 status of the tumour, it is important that a well-validated and robust methodology is chosen to minimise false negative or false positive determinations.
Immune-mediated adverse reactions.
Most immune-mediated adverse reactions occurring during treatment with atezolizumab were reversible with interruptions of atezolizumab and initiation of corticosteroids and/or supportive care. Immune-mediated adverse reactions affecting more than one body system have been observed. Immune-mediated adverse reactions with atezolizumab may occur after the last dose of atezolizumab. For suspected immune-mediated adverse reactions, a thorough evaluation to confirm aetiology or exclude other causes should be performed. Based on the severity of the adverse reaction, atezolizumab should be withheld and corticosteroids administered. Upon improvement to Grade ≤ 1, corticosteroids should be tapered over ≥ 1 month. Based on limited data from clinical studies in patients whose immune-mediated adverse reactions could not be controlled with systemic corticosteroid use, administration of other systemic immunosuppressants may be considered.
Tecentriq SC must be permanently discontinued for any Grade 3 immune-mediated adverse reaction that recurs; for any Grade 4 immune-mediated adverse reactions (except for endocrinopathies that are controlled with replacement hormones); and for some Grade 2 and 3 immune-mediated adverse reactions (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects)).
Immune-mediated pneumonitis.
Cases of pneumonitis, including fatal cases, have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of pneumonitis.
Treatment with Tecentriq SC should be withheld for Grade 2 pneumonitis, and 1 to 2 mg/kg prednisone or equivalent per day should be started. If symptoms improve to ≤ Grade 1, taper corticosteroids over ≥ 1 month. Treatment with Tecentriq SC may be resumed if the event improves to ≤ Grade 1 within 12 weeks, and corticosteroids have been reduced to ≤ 10 mg prednisone or equivalent per day. Treatment with Tecentriq SC must be permanently discontinued for Grade 3 or 4 pneumonitis.
Immune-mediated hepatitis.
Cases of hepatitis, some leading to fatal outcomes, have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of hepatitis. Monitor aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin prior to and periodically during treatment with Tecentriq SC. Appropriate management of patients with abnormal liver function tests (LFTs) at baseline should be considered.
Treatment with Tecentriq SC should be withheld if Grade 2 (ALT or AST > 3 to 5 x ULN or blood bilirubin > 1.5 to 3.0 x ULN) persists for more than 5 to 7 days, and 1 to 2 mg/kg prednisone or equivalent per day should be started. If the event improves to ≤ Grade 1, taper corticosteroids over ≥ 1 month.
Treatment with Tecentriq SC may be resumed if the event improves to ≤ Grade 1 within 12 weeks, and corticosteroids have been reduced to ≤ 10 mg oral prednisone or equivalent per day. Treatment with Tecentriq SC must be permanently discontinued for Grade 3 or Grade 4 events (ALT or AST > 5.0 x ULN or blood bilirubin > 3 x ULN).
Immune-mediated colitis.
Cases of diarrhoea or colitis have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of colitis. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis.
Treatment with Tecentriq SC should be withheld for Grade 2 or 3 diarrhoea (increase of ≥ 4 stools/day over baseline) or colitis (symptomatic). For Grade 2 diarrhoea or colitis, if symptoms persist > 5 days or recur, start 1-2 mg/kg prednisone or equivalent per day. Treat Grade 3 diarrhoea or colitis with IV corticosteroids (1 to 2 mg/kg/day methylprednisolone or equivalent). Once symptoms improve, treatment with 1 to 2 mg/kg/day of prednisone or equivalent should be started. If symptoms improve to ≤ Grade 1, taper corticosteroids over ≥ 1 month. Treatment with Tecentriq SC may be resumed if the event improves to ≤ Grade 1 within 12 weeks and corticosteroids have been reduced to ≤ 10 mg oral prednisone or equivalent per day. Treatment with Tecentriq SC must be permanently discontinued for Grade 4 (life threatening; urgent intervention indicated) diarrhoea or colitis.
Immune-mediated endocrinopathies.
Hypothyroidism, hyperthyroidism, adrenal insufficiency, hypophysitis and type 1 diabetes mellitus, including diabetic ketoacidosis, have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients should be monitored for clinical signs and symptoms of endocrinopathies and for changes in thyroid function (at the start of treatment, periodically during treatment and as indicated based on clinical evaluation). Patients may present with the following: fatigue, headache, mental status changes, heat or cold intolerance, tachycardia or bradycardia, unusual bowel habits, weight change, polyuria/polydipsia, blurred vision. Unless an alternative aetiology has been identified, signs and symptoms of endocrinopathies should be conservatively considered immune-mediated. Appropriate management of patients with abnormal thyroid function tests at baseline should be considered.
Asymptomatic patients with abnormal thyroid function tests can receive Tecentriq SC. For symptomatic hypothyroidism, Tecentriq SC should be withheld and thyroid hormone replacement should be initiated as needed. Isolated hypothyroidism may be managed with replacement therapy and without corticosteroids. For symptomatic hyperthyroidism, Tecentriq SC should be withheld and an anti-thyroid drug should be initiated as needed. Treatment with a beta blocker may also be considered. Treatment with Tecentriq SC may be resumed when symptoms are controlled and thyroid function is improving.
For symptomatic adrenal insufficiency, Tecentriq SC should be withheld and treatment with intravenous corticosteroids (1 to 2 mg/kg per day of methylprednisolone or equivalent) should be started. Once symptoms improve, follow with 1 to 2 mg/kg per day of prednisone or equivalent. If symptoms improve to ≤ Grade 1, taper corticosteroids over ≥ 1 month. Treatment may be resumed if the event improves to ≤ Grade 1 within 12 weeks and corticosteroids have been reduced to the equivalent of ≤ 10 mg prednisone or equivalent per day and the patient is stable on replacement therapy (if required).
Treatment with Tecentriq SC should be withheld for Grade 2 or Grade 3 hypophysitis. Treatment with intravenous corticosteroids (1 to 2 mg/kg per day IV methylprednisolone or equivalent) should be started, and hormone replacement should be initiated as needed. Once symptoms improve, treatment with 1 to 2 mg/kg per day of prednisone or equivalent should follow. If symptoms improve to ≤ Grade 1, corticosteroids should be tapered over ≥ 1 month. Treatment may be resumed if the event improves to ≤ Grade 1 within 12 weeks and corticosteroids have been reduced to ≤ 10 mg per day prednisone or equivalent and the patient is stable on replacement therapy (if required). Treatment with Tecentriq SC should be permanently discontinued for Grade 4 hypophysitis.
Treatment with insulin should be initiated for type 1 diabetes mellitus. For ≥ Grade 3 hyperglycaemia (fasting glucose greater than 13.9 mmol/L), Tecentriq SC should be withheld. Treatment with Tecentriq SC may be resumed if metabolic control is achieved on insulin replacement therapy.
Immune-mediated meningoencephalitis.
Meningoencephalitis has been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for clinical signs and symptoms of meningitis or encephalitis.
Treatment with Tecentriq SC must be permanently discontinued for any grade of meningitis or encephalitis. Treatment with intravenous corticosteroids (1 to 2 mg/kg IV methylprednisolone or equivalent per day) should be started. Once symptoms improve, treatment with 1 to 2 mg/kg oral prednisone or equivalent per day should follow.
Immune-mediated neuropathies.
Myasthenic syndrome/myasthenia gravis or Guillain-Barré syndrome, which may be life-threatening, and facial paresis were observed in patients receiving atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for symptoms of motor and sensory neuropathy.
Treatment with Tecentriq SC must be permanently discontinued for any grade of myasthenic syndrome/myasthenia gravis or Guillain-Barré syndrome. Consider initiation of systemic corticosteroids at a dose of 1 to 2 mg/kg oral prednisone or equivalent per day.
Treatment with Tecentriq SC must be permanently discontinued for Grade 3 or 4 facial paresis. Treatment with Tecentriq SC should be withheld for Grade 1 or 2 facial paresis, and treatment with systemic corticosteroids (1 to 2 mg/kg/day prednisone or equivalent) should be considered. Treatment may be resumed when symptoms improve to Grade 0 or Grade 1 within 12 weeks, and corticosteroids have been reduced to ≤ 10 mg prednisone or equivalent per day.
Immune-mediated myelitis.
Myelitis has been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be closely monitored for signs and symptoms that are suggestive of myelitis. Treatment with Tecentriq SC must be permanently discontinued for Grade 2, 3 or 4 myelitis.
Immune-mediated pancreatitis.
Pancreatitis, including increases in serum amylase and lipase levels, has been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be closely monitored for signs and symptoms that are suggestive of acute pancreatitis.
Treatment with Tecentriq SC should be withheld for ≥ Grade 3 serum amylase or lipase levels increased (> 2.0 ULN), or Grade 2 or 3 pancreatitis, and treatment with intravenous corticosteroids (1 to 2 mg/kg methylprednisolone or equivalent per day), should be started. Once symptoms improve, follow with 1 to 2 mg/kg oral prednisone or equivalent per day. Treatment with Tecentriq SC may be resumed when serum amylase and lipase levels improve to ≤ Grade 1 within 12 weeks, or symptoms of pancreatitis have resolved, and corticosteroids have been reduced to ≤ 10 mg prednisone or equivalent per day. Treatment with Tecentriq SC should be permanently discontinued for Grade 4, or any grade of recurrent pancreatitis.
Haemophagocytic lymphohistiocytosis.
Haemophagocytic lymphohistiocytosis (HLH), including fatal cases, has been reported in patients receiving atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). HLH should be considered when the presentation of cytokine release syndrome is atypical or prolonged. Patients should be monitored for clinical signs and symptoms of HLH. Treatment with Tecentriq SC must be permanently discontinued for suspected haemophagocytic lymphohistiocytosis.
Immune-mediated myocarditis.
Myocarditis, including fatal cases, has been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of myocarditis. Myocarditis may also be a clinical manifestation of myositis and should be managed accordingly.
Treatment with Tecentriq SC should be withheld for suspected myocarditis and treatment with systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or equivalent should be started. Treatment with Tecentriq SC must be permanently discontinued for Grade 2 or above myocarditis.
Immune-mediated myositis.
Cases of myositis, including fatal cases, have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs and symptoms of myositis. Patients with possible myositis should be monitored for signs of myocarditis.
Treatment with Tecentriq SC should be withheld for Grade 2 or 3 myositis and corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) should be initiated. If symptoms improve to ≤ Grade 1, taper corticosteroids as clinically indicated. Treatment with Tecentriq SC may be resumed if the event improves to ≤ Grade 1 within 12 weeks, and corticosteroids have been reduced to ≤ 10 mg prednisone or equivalent per day. Treatment with Tecentriq SC should be permanently discontinued for Grade 4 or Grade 3 recurrent myositis.
Immune-mediated pericardial disorders.
Pericardial disorders, including pericarditis, pericardial effusion and cardiac tamponade, some leading to fatal outcomes, have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects), Postmarketing experience). Patients should be monitored for clinical signs and symptoms of pericardial disorders.
For suspected Grade 1 pericarditis, treatment with Tecentriq SC should be withheld and prompt cardiology consultation with diagnostic workup according to current clinical guidelines should be initiated. For suspected Grade 2, 3 or 4 pericardial disorders, treatment with Tecentriq SC should be withheld, prompt treatment with systemic corticosteroids at a dose of 1 to 2 mg/kg/day of prednisone or equivalent should be started and prompt cardiology consultation with diagnostic workup according to current clinical guidelines should be initiated. Once a diagnosis of a pericardial disorder event is established, treatment with Tecentriq SC must be permanently discontinued for Grade 2, 3 or 4 pericardial disorders.
Immune-mediated nephritis.
Nephritis has been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for changes in renal function.
Treatment with Tecentriq SC should be withheld for Grade 2 nephritis. Treatment with systemic corticosteroids (1 to 2 mg/kg/day prednisone or equivalent) should be initiated. Treatment with Tecentriq SC may be resumed when the symptoms improve to Grade 0 or Grade 1 within 12 weeks and corticosteroids have been reduced to the equivalent of ≤ 10 mg prednisone or equivalent per day. Tecentriq SC must be permanently discontinued for Grade 3 or 4 nephritis.
Immune-mediated severe cutaneous adverse reactions.
Immune-mediated severe cutaneous adverse reactions (SCARs), including cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), have been reported in patients receiving atezolizumab. Patients should be monitored for suspected severe skin reactions and other causes should be excluded. Based on the severity of the adverse reaction, Tecentriq SC should be withheld for Grade 3 skin reactions and treatment with systemic corticosteroids at a dose of 1 - 2 mg/kg/day of prednisone or equivalent should be initiated. Treatment with Tecentriq SC may be resumed if the event improves to ≤ Grade 1 within 12 weeks, and corticosteroids have been reduced to ≤ 10 mg prednisone or equivalent per day. Treatment with Tecentriq SC should be permanently discontinued for Grade 4 skin reactions, and corticosteroids should be administered at a dose of 1 - 2 mg/kg/day of prednisone or equivalent.
For suspected SCARs, patients should be referred to a specialist for further diagnosis and management. Tecentriq SC should be withheld for patients with suspected SJS or TEN. For confirmed SJS or TEN, Tecentriq SC should be permanently discontinued.
Caution should be used when considering the use of Tecentriq SC in a patient who has previously experienced a severe or life-threatening skin adverse reaction on prior treatment with other immune-stimulatory anticancer agents.
Autoimmune haemolytic anaemia.
Atezolizumab can cause autoimmune haemolytic anaemia (AIHA). Patients should be monitored for signs and symptoms of drug-induced AIHA, and if this adverse reaction is observed, administration of Tecentriq SC should be permanently discontinued. Treatment for AIHA should be initiated, as deemed medically appropriate.
Infusion-related reactions.
Infusion-related reactions (IRRs), including hypersensitivity and anaphylaxis, have been observed in clinical trials with atezolizumab (see Section 4.8 Adverse Effects (Undesirable Effects)).
The rate of injection should be reduced or treatment should be interrupted in patients with Grade 1 or 2 infusion-related reactions. Tecentriq SC should be permanently discontinued in patients with Grade 3 or 4 infusion-related reactions. Patients with Grade 1 or 2 infusion-related reactions may continue to receive Tecentriq SC with close monitoring; premedication with an antipyretic and antihistamines may be considered.
Other immune-mediated adverse reactions.
The following additional clinically significant, immune-mediated adverse reactions have been reported in clinical studies with atezolizumab: uveitis (see Section 4.8 Adverse Effects (Undesirable Effects)). See Section 4.2 Dose and Method of Administration for recommended dose modifications (follow dose modification recommendations for 'Other immune-mediated adverse reactions').
Cases of haemolytic anaemia and aplastic anaemia have been observed during treatment with immune checkpoint inhibitors. Patients should be monitored for signs and symptoms indicative of these immune-mediated adverse reactions.
Patients with pre-existing autoimmune disease (AID).
In patients with pre-existing autoimmune disease (AID), data from observational studies suggest that the risk of immune-mediated adverse reactions following immune checkpoint inhibitor therapy may be increased as compared with the risk in patients without pre-existing AID. In addition, flares of the underlying AID were frequent, but the majority were mild and manageable.
Disease-specific precautions.
Patients excluded from clinical trials.
Patients with the following conditions were excluded from clinical trials: history of autoimmune disease, history of pneumonitis, active brain metastasis, HIV, hepatitis B or hepatitis C infection. Patients who were administered a live, attenuated vaccine within 28 days prior to enrolment; systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medicinal products within 2 weeks prior to study entry were excluded from clinical trials.
Patients with a baseline performance status ≥ 2 were excluded (apart from Study GO29293 [IMvigor210] Cohort 1 that enrolled patients with cisplatin-ineligible urothelial carcinoma and allowed a baseline performance status ≥ 2) (see Section 5.1 Pharmacodynamic Properties).
Use of atezolizumab in combination with bevacizumab, paclitaxel and carboplatin in metastatic non-squamous NSCLC.
Physicians should carefully consider the combined risks of the four-drug regimen of Tecentriq SC, bevacizumab, paclitaxel, and carboplatin before initiating treatment (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients with NSCLC that had clear tumour infiltration into the thoracic great vessels or clear cavitation of pulmonary lesions, as seen on imaging, were excluded from the pivotal clinical study IMpower150 after several cases of fatal pulmonary haemorrhage were observed, which is a known risk factor of treatment with bevacizumab. In the absence of data, Tecentriq SC should be used with caution in these populations after careful evaluation of the balance of benefits and risks for the patient.
Use of atezolizumab in urothelial carcinoma for previously untreated patients who are considered cisplatin ineligible.
The baseline and prognostic disease characteristics of the IMvigor210 Cohort 1 study population were overall comparable to patients in the clinic who would be considered cisplatin ineligible but would be eligible for a carboplatin based combination chemotherapy. There are insufficient data for the subgroup of patients that would be unfit for any chemotherapy; therefore Tecentriq SC should be used with caution in these patients, after careful consideration of the potential balance of risks and benefits on an individual basis.
Use of atezolizumab in combination with bevacizumab in hepatocellular carcinoma.
Bleeding (including fatal events) is a known adverse reaction with bevacizumab. Serious bleeding events, including fatalities, have occurred in hepatocellular cancer patients treated with the combination of atezolizumab and bevacizumab.
There is lack of clinical data to support the combination of atezolizumab and bevacizumab in hepatocellular cancer patients with bleeding varices (including recent bleeds), untreated varices or varices at high risk of bleeding because these patients were excluded from treatment with intravenous Tecentriq and bevacizumab in the IMbrave150 pivotal study (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Carefully consider the risks of Tecentriq SC plus bevacizumab in patients with HCC before initiating treatment. Patients with HCC should be evaluated for the presence of varices and have varices treated as indicated within 6 months prior to initiating therapy with the combination of Tecentriq SC and bevacizumab.
Refer to the bevacizumab Product Information for full prescribing information on the risks of bleeding events.
Use in hepatic impairment.
Based on a population pharmacokinetic analysis, no dose adjustment is required for patients with mild or moderate hepatic impairment (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties). There are no data in patients with moderate or severe hepatic impairment.
Use in renal impairment.
Based on a population pharmacokinetic analysis, no dose adjustment is required in patients with mild or moderate renal impairment (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties). Data from patients with severe renal impairment are too limited to draw conclusions on this population.
Use in the elderly.
No overall differences in safety or efficacy were observed between patients ≥ 65 years of age and younger patients (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Paediatric use.
Tecentriq SC is not approved for use in patients under the age of 18 years. The safety and efficacy of Tecentriq SC in the population has not been established. An early phase study conducted in paediatric and young adult patients did not demonstrate clinical benefit of atezolizumab.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No formal pharmacokinetic drug-drug interaction studies have been conducted with atezolizumab. Since atezolizumab is cleared from the circulation through catabolism, no metabolic drug-drug interactions are expected.
The use of systemic corticosteroids or immunosuppressants before starting atezolizumab should be avoided because of their potential interference with the pharmacodynamic activity and efficacy of atezolizumab. However, systemic corticosteroids or other immunosuppressants can be used to treat immune-mediated adverse reactions after starting atezolizumab.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No fertility studies have been conducted with atezolizumab however assessment of the cynomolgus monkey male and female reproductive organs was included in the chronic toxicity study. Atezolizumab had an effect on menstrual cycles in all female monkeys in the 50 mg/kg/week dose group characterised by an irregular cycle pattern during the dosing phase and correlated with the lack of fresh corpora lutea in the ovaries at the terminal necropsy; this effect was reversible during the dose-free recovery period. The AUC at the no effect level (15 mg/kg/week) was approximately 3.5 times that anticipated in patients at the clinical dose yielding the highest exposure. There was no effect on the male reproductive organs. No fertility studies were conducted with vorhyaluronidase alfa.
(Category D)
Based on the mechanism of action, the use of Tecentriq SC may cause fetal harm. Administration of Tecentriq SC is expected to have an adverse effect on pregnancy and poses a risk to the human fetus, including embryofetal lethality. Animal studies have demonstrated that inhibition of the PD-L1/PD-1 pathway can lead to an increased risk of immune-mediated rejection of the developing fetus resulting in fetal death.
No dedicated reproductive or teratogenicity studies in animals have been conducted with atezolizumab.
Tecentriq SC contains vorhyaluronidase alfa (see Section 6.1 List of Excipients). Reproductive toxicology studies with vorhyaluronidase alfa revealed embryofetal toxicity in mice at high systemic exposure, but did not show teratogenic potential.
There are no clinical studies of atezolizumab in pregnant women. Tecentriq SC is not recommended during pregnancy unless the potential benefit for the mother outweighs the potential risk to the fetus. Pregnant women should be advised of the potential risk to the fetus.
Women of childbearing potential should use highly effective contraception during treatment with Tecentriq SC and for 5 months after the last dose.
The safety of atezolizumab during labour and delivery has not been established.
It is not known whether atezolizumab is excreted in human breast milk. No studies have been conducted to assess the impact of atezolizumab on milk production or its presence in breast milk. As the potential for harm to the nursing infant is unknown, a decision must be made to either discontinue breast-feeding or discontinue Tecentriq SC therapy.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and to use machines have been performed.
Atezolizumb has minor influence on the ability to drive and use machines. Patients experiencing fatigue should be advised not to drive and use machines until symptoms abate.
4.8 Adverse Effects (Undesirable Effects)
The following categories of frequency have been used: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), very rare (< 1/10,000).
Atezolizumab monotherapy.
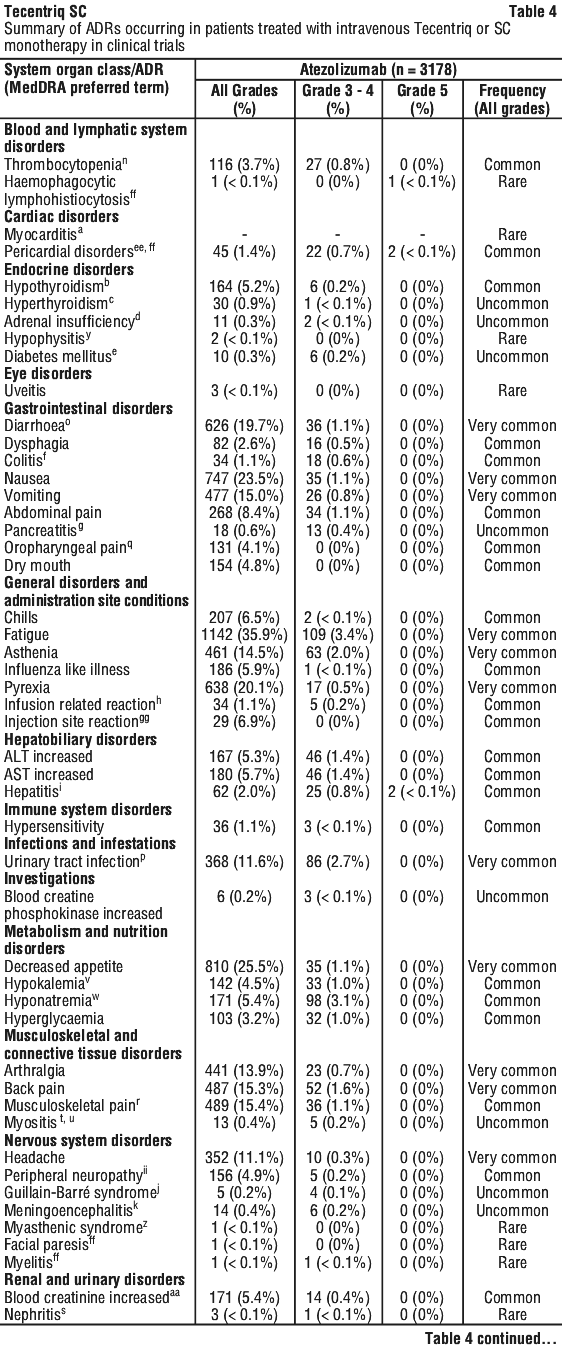
The safety of atezolizumab as a monotherapy is based on pooled data in 3178 patients across multiple tumour types with supporting data from the estimated cumulative exposure in > 13,000 patients across all clinical trials. The most common adverse reactions (> 10%) were fatigue (35.9%), decreased appetite (25.5%), nausea (23.5%), cough (20.8%), dyspnoea (20.5%), pyrexia (20.1%), diarrhoea (19.7%), rash (19.3%), musculoskeletal pain (15.4%), back pain (15.3%), vomiting (15.0%), asthenia (14.5%), arthralgia (13.9%), pruritus (12.6%), headache (11.1%) and urinary tract infection (11.6%).
Table 4 summarises the adverse drug reactions (ADRs) that have been reported in association with the use of intravenous Tecentriq or Tecentriq SC monotherapy.

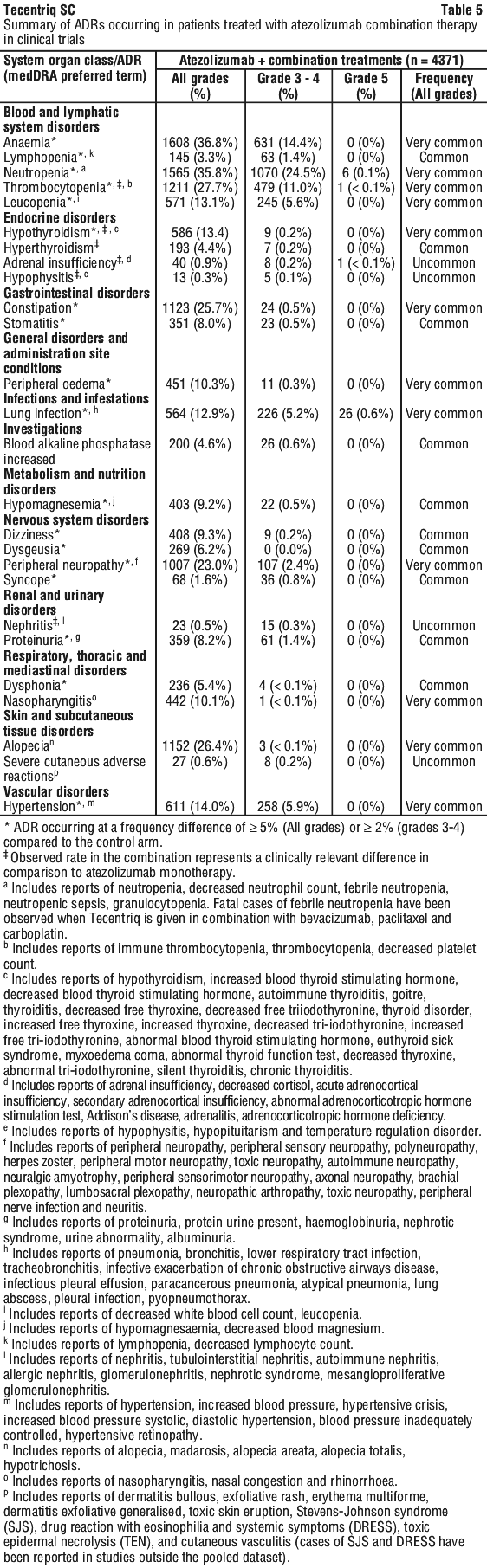
Atezolizumab combination therapy.
The safety of atezolizumab given in combination with other medicinal products is based on pooled data in 4,371 patients in clinical trials across multiple tumour types. Additional ADRs associated with the use of atezolizumab in combination therapy (not reported in monotherapy trials) are summarised in Table 5. ADRs with a clinically relevant difference when compared to monotherapy (see Table 4) are also presented. The most common adverse reactions (≥ 10%) were anaemia (36.8%), neutropenia (35.8%), thrombocytopenia (27.7%), alopecia (26.4%), constipation (25.7%), peripheral neuropathy (23.0%), hypertension (14.0%), hypothyroidism (13.4%), leucopenia (13.1%), lung infection (12.9%), peripheral oedema (10.3%) and nasopharyngitis (10.1%).
 One patient treated with intravenous Tecentriq in combination with carboplatin and etoposide in Study GO30081 (IMpower133) experienced Grade 3 anaphylaxis and discontinued treatment with Tecentriq.
One patient treated with intravenous Tecentriq in combination with carboplatin and etoposide in Study GO30081 (IMpower133) experienced Grade 3 anaphylaxis and discontinued treatment with Tecentriq.
Additional information for selected adverse reactions.
The data below reflect information for significant adverse reactions for atezolizumab monotherapy. Details for the significant adverse reactions for atezolizumab when given in combination are presented if clinically relevant differences were noted in comparison to atezolizumab monotherapy. See Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use for management of the following:
Immune-mediated pneumonitis. Pneumonitis occurred in 2.7% (87/3178) of patients who received atezolizumab monotherapy. Of the 87 patients, one event was fatal. The median time to onset was 3.4 months (range: 0.1 to 24.8 months). The median duration was 1.4 months (range 0 to 21.2+ months; + denotes a censored value). Pneumonitis led to discontinuation of atezolizumab in 12 (0.4%) patients. Pneumonitis requiring the use of corticosteroids occurred in 1.6% (51/3178) of patients receiving atezolizumab.
Immune-mediated hepatitis. Hepatitis occurred in 2.0% (62/3178) of patients who received atezolizumab monotherapy. Of the 62 patients, two events were fatal. The median time to onset was 1.5 months (range 0.2 to 18.8 months). The median duration was 2.1 months (range 0 to 22.0+ months; + denotes a censored value). Hepatitis led to discontinuation of atezolizumab in 6 (0.2%) patients. Hepatitis requiring the use of corticosteroids occurred in 0.6% (18/3178) of patients receiving atezolizumab.
Immune-mediated colitis. Colitis occurred in 1.1% (34/3178) of patients who received atezolizumab. The median time to onset was 4.7 months (range 0.5 to 17.2 months). The median duration was 1.2 months (range 0.1 to 17.8+ months; + denotes a censored value). Colitis led to discontinuation of atezolizumab in 8 (0.3%) patients. Colitis requiring the use of corticosteroids occurred in 0.6% (19/3178) of patients receiving atezolizumab.
Immune-mediated endocrinopathies. Thyroid disorders.
Hypothyroidism occurred in 5.2% (164/3178) of patients who received atezolizumab monotherapy. The median time to onset was 4.9 months (range 0 to 31.3 months).
Hyperthyroidism occurred in 0.9% (30/3178) of patients who received atezolizumab monotherapy. The median time to onset was 2.1 months (range 0.7 to 15.7 months). The median duration was 2.6 months (range: 0+ to 17.1+ months; + denotes a censored value).
Hyperthyroidism occurred in 4.9% (23/473) of patients who received atezolizumab in combination with carboplatin and nab-paclitaxel. Hyperthyroidism led to discontinuation in 1 (0.2%) patient.
Adrenal insufficiency.
Adrenal insufficiency occurred in 0.3% (11/3178) of patients who received atezolizumab monotherapy. The median time to onset was 5.5 months (range 0.1 to 19.0 months). The median duration was 16.8 months (range 0 to 16.8 months). Adrenal insufficiency led to discontinuation of atezolizumab in 1 (< 0.1%) patient. Adrenal insufficiency requiring the use of corticosteroids occurred in 0.3% (9/3178) of patients receiving atezolizumab.
Adrenal insufficiency occurred in 1.5% (7/473) of patients who received atezolizumab in combination with carboplatin and nab-paclitaxel. Adrenal insufficiency requiring the use of corticosteroids occurred in 0.8% (4/473) of patients receiving atezolizumab in combination with carboplatin and nab-paclitaxel.
Hypophysitis.
Hypophysitis occurred in < 0.1% (2/3178) of patients who received atezolizumab monotherapy. The median time to onset was 7.2 months (range: 0.8 to 13.7 months). One patient required the use of corticosteroids and treatment with atezolizumab was discontinued.
Hypophysitis occurred in 0.8% (3/393) of patients who received atezolizumab with bevacizumab, paclitaxel and carboplatin. The median time to onset was 7.7 months (range: 5.0 to 8.8 months). Two patients required the use of corticosteroids. Hypophysitis led to the discontinuation of treatment in one patient.
Diabetes mellitus.
Diabetes mellitus occurred in 0.3% (10/3178) of patients who received atezolizumab monotherapy. The median time to onset was 4.2 months (range 0.1 to 9.9 months). The median duration was 1.6 months (range: 0.1 to 15.2+ months; + denotes a censored value). Diabetes mellitus led to the discontinuation of atezolizumab in 3 (< 0.1%) patients.
Immune-mediated meningoencephalitis. Meningoencephalitis occurred in 0.4% (14/3178) of patients who received atezolizumab monotherapy. The median time to onset was 0.5 months (range 0 to 12.5 months). The median duration was 0.7 months (range 0.2 to 14.5+ months; + denotes a censored value). Meningoencephalitis requiring the use of corticosteroids occurred in 0.2% (6/3178) of patients receiving atezolizumab and led to discontinuation of atezolizumab in 4 (0.1%) patients.
Immune-mediated neuropathies. Guillain-Barré syndrome and demyelinating polyneuropathy.
Guillain-Barré syndrome and demyelinating polyneuropathy, occurred in 0.2% (5/3178) of patients who received atezolizumab monotherapy. The median time to onset was 7.0 months (range: 0.6 to 8.1 months). The median duration was 8.0 months (0.6 to 8.3+ months; + denotes a censored value). Guillain-Barré syndrome led to the discontinuation of atezolizumab in 1 (< 0.1%) patient. Guillain-Barré syndrome requiring the use of corticosteroids occurred in < 0.1% (2/3178) of patients receiving atezolizumab.
Facial paresis.
Facial paresis occurred in < 0.1% (1/3178) of patients who received atezolizumab monotherapy. The time to onset was 0.95 months. The duration was 1.1 months. The event did not require the use of corticosteroids and the event did not lead to the discontinuation of atezolizumab.
Immune-mediated myelitis. Myelitis occurred in < 0.1% (1/3178) of patients who received atezolizumab monotherapy. The time to onset was 0.76 months. The event required the use of corticosteroids but did not lead to the discontinuation of atezolizumab.
Immune-mediated pancreatitis. Pancreatitis, including amylase increased and lipase increased, occurred in 0.6% (18/3178) of patients who received atezolizumab monotherapy. The median time to onset was 5.0 months (range 0.3 to 16.9 months). The median duration was 0.8 months (range 0.1 to 12.0+ months; + denotes a censored value). Pancreatitis led to discontinuation of atezolizumab in 3 (< 0.1%) patients. Pancreatitis requiring the use of corticosteroids occurred in 0.1% (4/3178) of patients receiving atezolizumab.
Haemophagocytic lymphohistiocytosis. Haemophagocytic lymphohistiocytosis (HLH) occurred in < 0.1% (1/3178) of patients who received atezolizumab monotherapy. The time to onset was 1.6 months. The duration was 1.4 months. HLH led to discontinuation of atezolizumab in 1 (< 0.1%) patient. The patient did not require the use of corticosteroids.
Immune-mediated myocarditis. Myocarditis occurred in < 0.1% (2/8000) of patients across all atezolizumab clinical trials in multiple tumour types and treatment combinations. The time to onset was 18 and 33 days. Both patients required corticosteroids and discontinued atezolizumab.
Immune-mediated pericardial disorders. Pericardial disorders occurred in 1.4% (45/3178) of patients who received atezolizumab monotherapy. The median time to onset was 1.4 months (range 0.2 to 17.5 months). The median duration was 1.4 months (range 0 to 19.3 months). Pericardial disorders led to discontinuation of atezolizumab in 3 (< 0.1%) patients. Pericardial disorders requiring the use of corticosteroids occurred in 0.2% (7/3178) of patients.
Immune-mediated myositis. Myositis occurred in 0.4% (13/3178) of patients who received atezolizumab monotherapy. The median time to onset was 5.1 months (range 0.7 to 11.0 months). The median duration was 5.0 months (range 0.7 to 22.6+ months, + denotes a censored value). Myositis led to discontinuation of atezolizumab in 1 (< 0.1%) patient. Seven (0.2%) patients required the use of corticosteroids.
Immune-mediated nephritis. Nephritis occurred in < 0.1% (3/3178) of patients who received atezolizumab monotherapy. The median time to onset was 13.1 months (range 9.0 to 17.5 months). The median duration was 2.8 months (range 0.5 to 9.5+ months, + denotes a censored value). Nephritis led to discontinuation of atezolizumab in 2 (< 0.1%) of patients. One patient required the use of corticosteroids.
Immune-mediated severe cutaneous adverse reactions. Severe cutaneous adverse reactions (SCARs) occurred in 0.7% (22/3178) of patients who received atezolizumab monotherapy. The median time to onset was 5.9 months (range 0.1 to 15.5 months). The median duration was 1.6 months (range 0 to 22.1+ months; + denotes a censored value). SCARs led to discontinuation of atezolizumab in 3 (< 0.1%) patients. SCARs requiring the use of systemic corticosteroids occurred in 0.2% (6/3178) of patients receiving atezolizumab monotherapy.
Use of Tecentriq in combination with bevacizumab, paclitaxel and carboplatin. In Study GO29436 (IMpower150), an overall higher frequency of adverse events was observed in the four-drug regimen of intravenous Tecentriq, bevacizumab, paclitaxel, and carboplatin compared to intravenous Tecentriq, paclitaxel and carboplatin, including Grade 3 and 4 events (63.6% compared to 57.5%), Grade 5 events (6.1% compared to 2.5%), adverse events of special interest to intravenous Tecentriq (52.4% compared to 48.0%), as well as adverse events leading to withdrawal of any study treatment (33.8% compared to 13.3%). Nausea, diarrhoea, stomatitis, fatigue, pyrexia, mucosal inflammation, decreased appetite, weight decreased, hypertension and proteinuria were reported higher (≥ 5% difference) in patients receiving intravenous Tecentriq in combination with bevacizumab, paclitaxel and carboplatin. Other clinically significant adverse events which were observed more frequently in the intravenous Tecentriq, bevacizumab, paclitaxel, and carboplatin arm were epistaxis, haemoptysis, cerebrovascular accident, including fatal events.
Immune checkpoint inhibitor class effects. There have been cases of the following adverse reaction(s) reported during treatment with other immune checkpoint inhibitors which might also occur during treatment with atezolizumab: pancreatic exocrine insufficiency.
Immunogenicity.
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralising antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to atezolizumab with the incidence of antibodies to other products may be misleading.
Intravenous Tecentriq.
Across multiple phase II and III studies with intravenous atezolizumab, 13.1% to 54.1% of patients developed treatment-emergent anti-drug antibodies (ADAs) and 4.3% to 27.5% of patients developed neutralising antibodies (NAbs). The median time to ADA onset ranged from 3 weeks to 5 weeks.
A decrease in exposure (9% increase in clearance) was observed in ADA-positive patients compared to ADA-negative patients; however, this effect on exposure is not expected to be clinically meaningful given the flat exposure-response relationship and adequate target exposure achieved regardless of ADA status.
Patients who developed treatment emergent ADAs tended to have overall poorer health and disease characteristics at baseline. Exploratory analyses adjusting for imbalances in baseline health and disease characteristics were conducted to assess the effect of ADA on efficacy. These analyses did not exclude possible attenuation of efficacy benefit in patients who develop ADA compared to patients who did not develop ADA.
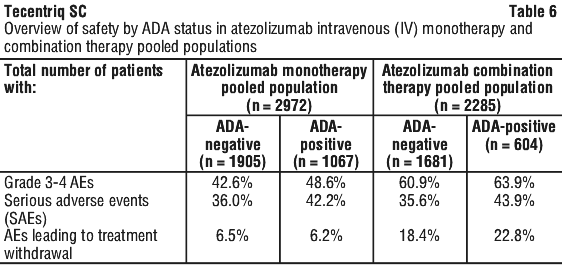
Across pooled datasets for patients treated with atezolizumab monotherapy and with combination therapies, the rates of adverse events (AEs) which have been observed for the ADA-positive population compared to the ADA-negative population is presented in Table 6. Available data do not allow conclusions to be drawn on possible patterns of adverse drug reactions or their causal relationship with ADAs.
Tecentriq SC.
In IMscin001, the incidence of treatment-emergent anti-atezolizumab antibodies in patients treated with Tecentriq SC and intravenous Tecentriq was comparable (19.5% [43/221] and 13.9% [15/108], respectively). Anti-atezolizumab antibody status did not appear to have a clinically relevant impact on atezolizumab PK, efficacy or safety. The incidence of treatment-emergent anti-rHuPH20 (vorhyaluronidase alfa) antibodies in patients treated with Tecentriq SC was 5.4% (12/224). The clinical relevance of the development of anti-rHuPH20 antibodies after treatment with Tecentriq SC is unknown.
Laboratory abnormalities.
All identified laboratory abnormalities were reported as ADRs. See Section 4.4 Special Warnings and Precautions for Use, Immune-mediated hepatitis and Immune-mediated endocrinopathies for management of the following: AST, ALT, bilirubin; thyroid function.
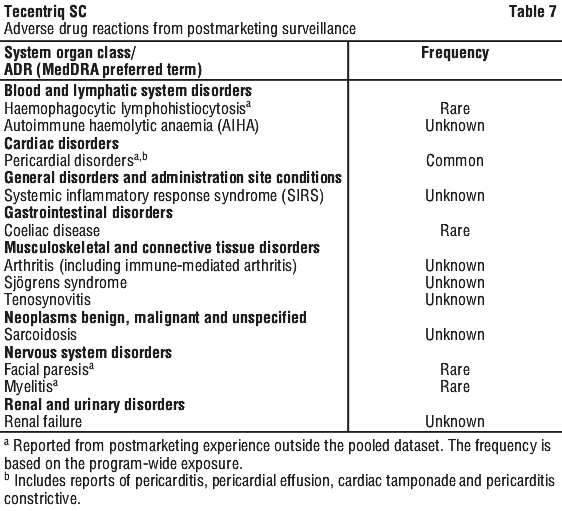
Postmarketing experience.
The following adverse reactions have been identified during post-approval use of atezolizumab (see Table 7). Because reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Reporting of suspected adverse reactions.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no information on overdose with Tecentriq SC.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
ATC code: L01FF05.
Mechanism of action.
Binding of PD-L1 to the PD-1 and B7.1 receptors found on T cells suppresses cytotoxic T-cell activity through the inhibition of T-cell proliferation and cytokine production. PD-L1 may be expressed on tumour cells (TC) and tumour-infiltrating immune cells (IC), and can contribute to the inhibition of the anti-tumour immune response in the microenvironment.
Atezolizumab is an Fc-engineered humanised immunoglobulin G1 (IgG1) monoclonal antibody that directly binds to PD-L1 and blocks interactions with the PD-1 and B7.1 receptors, releasing PD-L1/PD-1 pathway-mediated inhibition of the immune response, including reactivating the anti-tumour immune response. Atezolizumab leaves the PD-L2/PD-1 interaction intact, allowing PD-L2/PD-1 mediated inhibitory signals to persist. In syngeneic mouse tumour models, blocking PD-L1 activity resulted in decreased tumour growth.
PD-L1 expression by immunohistochemistry.
In certain clinical studies with atezolizumab (see Clinical trials below), the VENTANA PD-L1 (SP263) Assay or the VENTANA PD-L1 (SP142) Assay was used in accordance with validated usage to detect PD-L1 expression: either in tumour cells (TC) only (SP263) or in both tumour-infiltrating immune cells (IC) and TC (SP142) (see Section 5.1 Pharmacodynamic Properties).
Clinical trials.
This section presents clinical experience from studies investigating the use of Tecentriq monotherapy or combination therapy in early-stage and metastatic NSCLC, SCLC, UC, and HCC. With the exception of IMscin001, all of the studies were conducted using intravenous Tecentriq. The use of Tecentriq SC for the indications studied in intravenous Tecentriq is based on the pharmacokinetic non-inferiority of subcutaneously administered atezolizumab to intravenously administered atezolizumb, as demonstrated in IMscin001 (see Section 5.2 Pharmacokinetic Properties).
Tecentriq SC.
IMscin001 (BP40657).
A phase Ib/III, open-label, multi-centre, international, randomised study, IMscin001, was conducted to evaluate the pharmacokinetics, efficacy and safety of Tecentriq SC compared with intravenous Tecentriq in patients with locally advanced or metastatic NSCLC who have not been exposed to cancer immunotherapy (CIT) and for whom prior platinum-based therapy has failed. IMscin001 was designed to demonstrate non-inferiority of the atezolizumab Cycle 1 (pre-dose Cycle 2) serum Ctrough and model-predicted AUC from 0 to 21 days at Cycle 1 of atezolizumab SC compared with intravenous atezolizumab (co-primary endpoint). Secondary endpoints included efficacy [progression free survival (PFS), objective response rate (ORR), overall survival (OS), duration of response (DOR)] and patient reported outcomes.
In Part 2 (Phase III), a total of 371 patients were enrolled and randomised to receive either 1875 mg of Tecentriq SC every 3 weeks or 1200 mg of intravenous Tecentriq every 3 weeks. No dose reduction was allowed.
Patients were excluded if they had a history of autoimmune disease; active or corticosteroid-dependent brain metastases, administration of a live, attenuated vaccine within 4 weeks prior to randomisation; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomisation.
The demographics and baseline disease characteristics were generally balanced between the treatment arms. The median age was 64 years (range: 27 to 85), and 69% of patients were male. The majority of patients were White (67%). Approximately two-thirds of patients (65%) had non-squamous disease, 5% had known EGFR mutation, 2% had known ALK rearrangements, 40% were PD-L1 positive (TC ≥ 1% and/or IC ≥ 1%), 16% had non-active CNS metastases at baseline, 26% had an ECOG PS of 0, 74% had an ECOG PS of 1, and most patients were current or previous smokers (70%). 80% received one prior therapeutic regimen.
Non-inferiority of the exposure from atezolizumab in Tecentriq SC compared to intravenous atezolizumab was demonstrated (see Section 5.2 Pharmacokinetic Properties). Other key results are summarised in Table 8. At the time of primary analysis, the median survival follow-up was 4.7 months and OS and DOR results were immature. There were 86 (35%) deaths in the Tecentriq SC arm and 37 (30%) deaths in the intravenous atezolizumab arm.

Patient experience.
IMscin002 (MO43576).
The IMscin002 study was a randomised, multi-centre, open-label cross-over trial conducted in patients with NSCLC with the primary objective to evaluate patient preference for Tecentriq SC compared with intravenous Tecentriq. The 179 patients randomised in the study had either PD-L1-positive early-stage NSCLC and completed adjuvant treatment or were chemotherapy-naïve with high PD-L1 stage IV NSCLC. Following randomisation, patients received 3 cycles of Tecentriq SC followed by 3 cycles of intravenous Tecentriq (Arm A) or 3 cycles of intravenous Tecentriq followed by 3 cycles of Tecentriq SC (Arm B).
Out of the 126 eligible patients, 123 (98%) completed the patient preference questionnaire (PPQ). At primary analysis, 87 out of 123 patients (71%) reported preferring subcutaneous administration of Tecentriq SC over intravenous Tecentriq and the main reason cited was that administration required less time in the clinic. Twenty-six out of 123 patients (21%) reported preferring intravenous Tecentriq over Tecentriq SC and the main reason cited was that it felt more comfortable during administration. Ten out of 123 patients (8%) had no preference for the route of administration.
Following the crossover periods, patients in both arms could continue treatment for up to 16 cycles (patients with early-stage NSCLC) or until disease progression or unacceptable toxicity (patients with stage IV NSCLC). Out of the 107 patients who entered the treatment continuation period, 85 (79%) patients (42 from intravenous/SC and 43 from SC/intravenous) chose to continue treatment with the SC route of administration.
Intravenous Tecentriq.
Non-small cell lung cancer. Early-stage NSCLC. IMpower010 (GO29527).
A phase III, open label, multi-centre, randomised study, GO29527 (IMpower010), was conducted to evaluate the efficacy and safety of Tecentriq for the adjuvant treatment of patients with stage IB (tumours ≥ 4 cm) - IIIA NSCLC (per the Union for International Cancer Control/American Joint Committee on Cancer staging system, 7th edition). A total of 1280 enrolled patients had complete tumour resection and were eligible to receive up to 4 cycles of cisplatin-based chemotherapy. The cisplatin-based chemotherapy regimens are described in Table 9.
 After completion of cisplatin-based chemotherapy (up to four cycles), a total of 1005 patients were randomised in a 1:1 ratio to receive Tecentriq (Arm A) or best supportive care (BSC) (Arm B). Tecentriq was administered as a fixed dose of 1200 mg by IV infusion every 3 weeks for 16 cycles unless there was disease recurrence or unacceptable toxicity. Randomisation was stratified by sex, stage of disease, histology, and PD-L1 expression.
After completion of cisplatin-based chemotherapy (up to four cycles), a total of 1005 patients were randomised in a 1:1 ratio to receive Tecentriq (Arm A) or best supportive care (BSC) (Arm B). Tecentriq was administered as a fixed dose of 1200 mg by IV infusion every 3 weeks for 16 cycles unless there was disease recurrence or unacceptable toxicity. Randomisation was stratified by sex, stage of disease, histology, and PD-L1 expression.
Patients were excluded if they had a history of autoimmune disease; administration of a live, attenuated vaccine within 28 days prior to randomisation; administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomisation. Tumour assessments were conducted at baseline of the randomisation phase and every 4 months for the first year following Cycle 1, Day 1 and then every 6 months until year five, then annually thereafter.
The median age was 62 years (range: 26 to 84), and 67% of patients were male. The majority of patients were White (73%), and 24% were Asian. Most patients were current or previous smokers (78%) and baseline ECOG performance status in patients was 0 (55%) or 1 (44%). Overall, 12% of patients had stage IB, 47% had stage II and 41% had stage IIIA disease. As measured by the VENTANA PD-L1 (SP263) Assay, 55% of patients had tumours with PD-L1 expression ≥ 1% on TC and 26% of patients had tumours with PD-L1 expression ≥ 50% on TC.
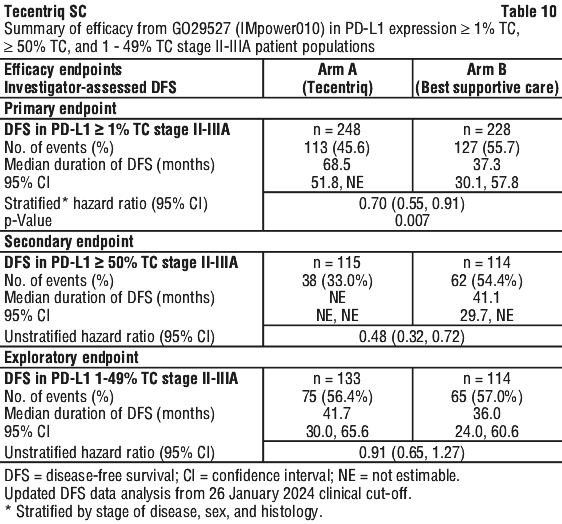
The primary efficacy outcome measure was disease-free survival (DFS) as assessed by the investigator. DFS was defined as the time from the date of randomisation to the date of occurrence of any of the following: first documented recurrence of disease, new primary NSCLC, or death due to any cause, whichever occurred first. DFS was assessed hierarchically in the following patient populations: stage II-IIIA NSCLC with PD-L1 expression ≥ 1% TC, all randomised patients with stage II-IIIA NSCLC, and all randomised ITT patients. DFS in the PD-L1 ≥ 50% TC stage II-IIIA population and OS in the ITT population were pre-specified key secondary objectives.
At the time of the interim DFS analysis, the study met its primary endpoint and demonstrated a statistically significant improvement in DFS in the Tecentriq arm compared to the BSC arm in the PD-L1 ≥ 1% TC stage II - IIIA patient population. The median follow-up time was approximately 32 months.
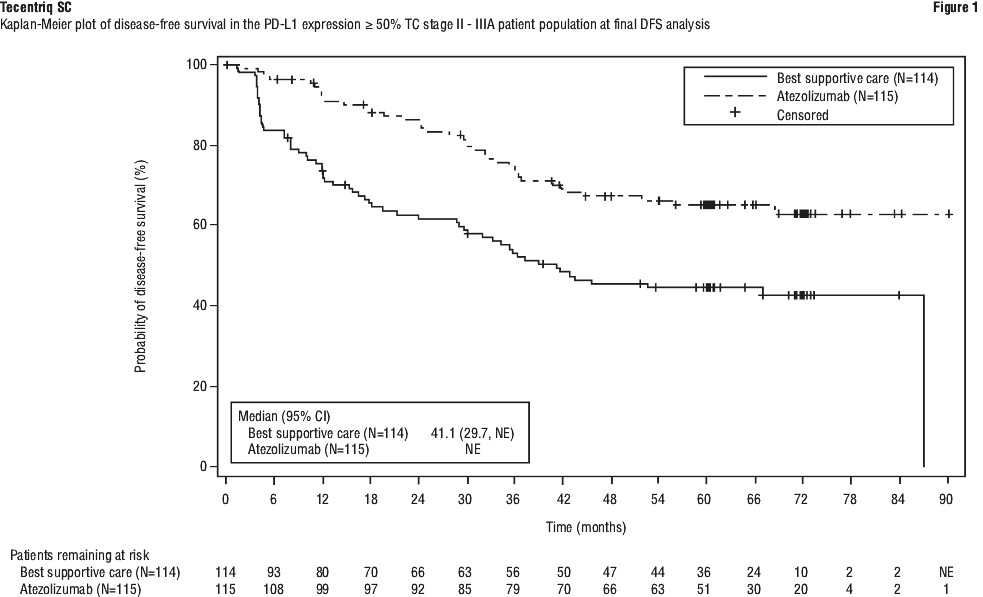
In the secondary objective analysis of stage II-IIIA patients with PD-L1 TC ≥ 50%, a clinically meaningful improvement in DFS was shown in the Tecentriq arm compared to the BSC arm with an unstratified HR of 0.43 (95% CI: 0.27, 0.68). Results were consistent at the time of the final DFS analysis, with median follow up time of 65 months. The OS data were immature at the time of the DFS final analysis.
The key efficacy results are summarised in Table 10. The Kaplan-Meier curve for DFS for the PD-L1 ≥ 50% TC stage II-IIIA patient population is presented in Figure 1.
 1L metastatic non-squamous NSCLC.
1L metastatic non-squamous NSCLC. IMpower150 (GO29436).
A phase III, open-label, multicentre, international, randomised study, IMpower150 (GO29436), was conducted to evaluate the efficacy and safety of Tecentriq in combination with paclitaxel and carboplatin, with or without bevacizumab, in chemotherapy-naïve patients with metastatic non-squamous NSCLC.
Patients were excluded if they had history of autoimmune disease, administration of a live, attenuated vaccine within 28 days prior to randomisation, administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomisation, active or untreated CNS metastases, clear tumour infiltration into the thoracic great vessels or clear cavitation of pulmonary lesions, as seen on imaging. Tumour assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter. Tumour specimens were evaluated for PD-L1 expression on tumour cells (TC) and tumour-infiltrating immune cells (IC) using the VENTANA PD-L1 (SP142) Assay and the results were used to define the PD-L1 expression subgroups for the analyses described below.
A total of 1202 patients were enrolled and were randomised (1:1:1) to receive one of the treatment regimens described in Table 11. Randomisation was stratified by sex, presence of liver metastases and PD-L1 tumour expression on TC and IC.
 The demographics and baseline disease characteristics of the study population were well balanced between the treatment arms. The median age was 63 years (range: 31 to 90), and 60% of patients were male. The majority of patients were White (82%). Approximately 10% of patients had known EGFR mutation, 4% had known ALK rearrangements, 14% had liver metastasis at baseline, and most patients were current or previous smokers (80%). Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (43%) or 1 (57%). 51% of patients' tumours had PD-L1 expression of ≥ 1% TC or ≥ 1% IC and 49% of patients' tumours had PD-L1 expression of < 1% TC and < 1% IC.
The demographics and baseline disease characteristics of the study population were well balanced between the treatment arms. The median age was 63 years (range: 31 to 90), and 60% of patients were male. The majority of patients were White (82%). Approximately 10% of patients had known EGFR mutation, 4% had known ALK rearrangements, 14% had liver metastasis at baseline, and most patients were current or previous smokers (80%). Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (43%) or 1 (57%). 51% of patients' tumours had PD-L1 expression of ≥ 1% TC or ≥ 1% IC and 49% of patients' tumours had PD-L1 expression of < 1% TC and < 1% IC.
At the time of the final analysis for PFS, patients had a median follow up time of 15.3 months. The ITT population, including patients with EGFR mutations or ALK rearrangements who should have been previously treated with tyrosine kinase inhibitors, demonstrated clinically meaningful PFS improvement in Arm B as compared to Arm C (HR of 0.61, 95% CI: 0.52, 0.72; median PFS 8.3 vs. 6.8 months).
At the time of the interim OS analysis, patients had a median follow-up of 19.7 months. The key results from this analysis as well as from the updated PFS analysis in the ITT population are summarised in Tables 12 and 13. The Kaplan-Meier curve for OS in the ITT population is presented in Figure 2. Figure 3 summarises the results of OS in the ITT and PD-L1 subgroups. Updated PFS results are also presented in Figures 4 and 5.
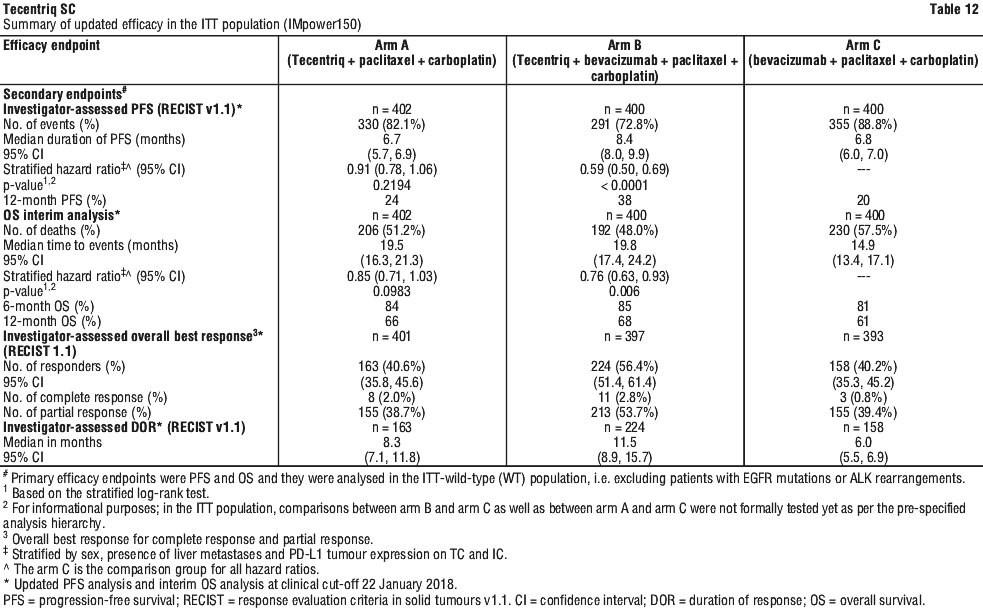
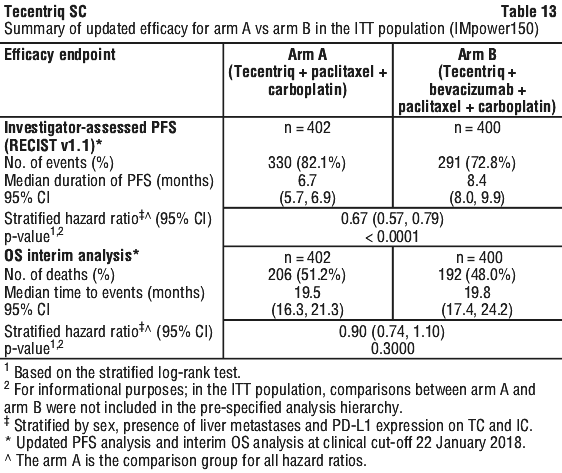


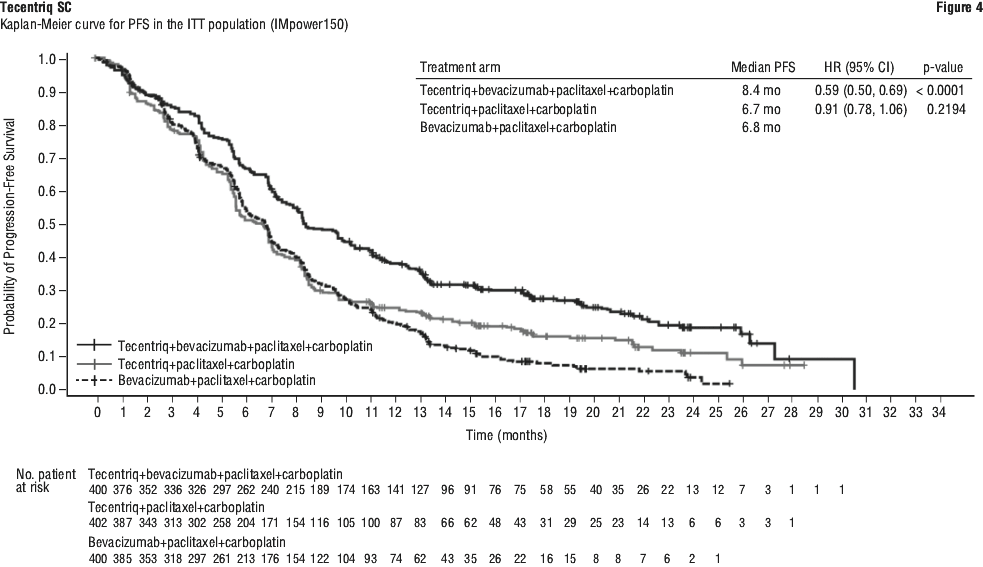
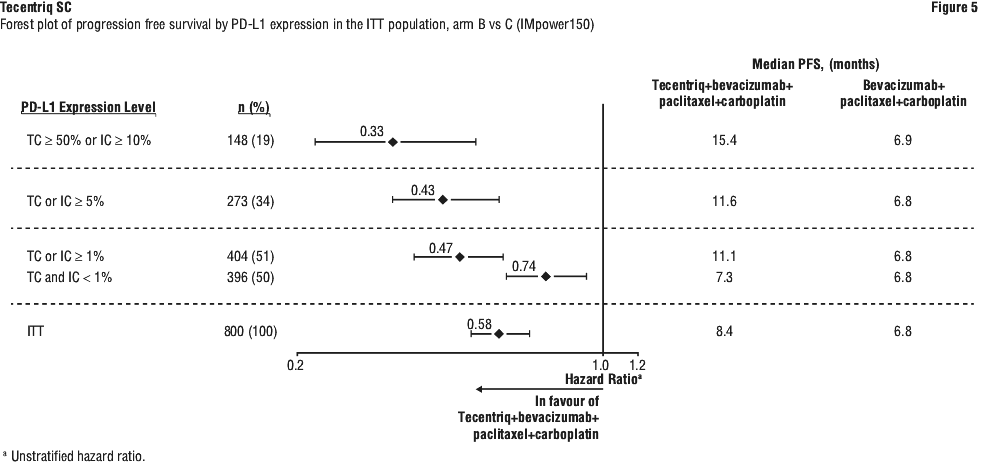 In Arm B as compared to Arm C, pre-specified subgroup analyses from the interim OS analysis showed an OS improvement for patients with EGFR mutations or ALK rearrangements (hazard ratio [HR] of 0.54, 95% CI: 0.29, 1.03; median OS not reached vs. 17.5 months), and liver metastases (HR of 0.52, 95% CI: 0.33, 0.82; median OS 13.3 vs 9.4 months). PFS improvements were also shown in patients with EGFR mutations or ALK rearrangements (HR of 0.55, 95% CI: 0.35, 0.87; median PFS 10.0 vs. 6.1 months) and liver metastases (HR of 0.41, 95% CI: 0.26, 0.62; median PFS 8.2 vs. 5.4 months). OS results were similar for patients aged < 65 and ≥ 65 subgroups, respectively. Data for patients ≥ 75 years of age are too limited to draw conclusions on this population. For all subgroup analyses, formal statistical testing was not planned.
In Arm B as compared to Arm C, pre-specified subgroup analyses from the interim OS analysis showed an OS improvement for patients with EGFR mutations or ALK rearrangements (hazard ratio [HR] of 0.54, 95% CI: 0.29, 1.03; median OS not reached vs. 17.5 months), and liver metastases (HR of 0.52, 95% CI: 0.33, 0.82; median OS 13.3 vs 9.4 months). PFS improvements were also shown in patients with EGFR mutations or ALK rearrangements (HR of 0.55, 95% CI: 0.35, 0.87; median PFS 10.0 vs. 6.1 months) and liver metastases (HR of 0.41, 95% CI: 0.26, 0.62; median PFS 8.2 vs. 5.4 months). OS results were similar for patients aged < 65 and ≥ 65 subgroups, respectively. Data for patients ≥ 75 years of age are too limited to draw conclusions on this population. For all subgroup analyses, formal statistical testing was not planned.
IMpower130 (GO29537).
A phase III, open-label, randomised study, IMpower130 (GO29537) was conducted to evaluate the efficacy and safety of Tecentriq in combination with nab-paclitaxel and carboplatin, in chemotherapy-naïve patients with metastatic non-squamous NSCLC. Patients including those with EGFR or ALK genomic tumour aberrations, were enrolled and were randomised in a 2:1 ratio to receive one of the treatment regimens described in Table 14. Randomisation was stratified by sex, presence of liver metastases and PD-L1 tumour expression on tumour cells (TC) and tumour infiltrating cells (IC) according to the VENTANA PD-L1 (SP142) Assay. Patients in treatment regimen B were able to crossover and receive Tecentriq monotherapy following disease progression.
 Patients were excluded if they had history of autoimmune disease, administration of live, attenuated vaccine within 28 days prior to randomisation, administration of immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomisation, and active or untreated CNS metastases. Tumour assessments were conducted every 6 weeks for the first 48 weeks following cycle 1, then every 9 weeks thereafter.
Patients were excluded if they had history of autoimmune disease, administration of live, attenuated vaccine within 28 days prior to randomisation, administration of immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomisation, and active or untreated CNS metastases. Tumour assessments were conducted every 6 weeks for the first 48 weeks following cycle 1, then every 9 weeks thereafter.
The demographics and baseline disease characteristics of the study population (n = 723) were well balanced between the treatment arms. The median age was 64 years (range 18 to 86). The majority of the patients were, male (57%), White (90%). 14.8% of patients had liver metastases at baseline, and most patients were current or previous smokers (88%). The majority of patients had ECOG performance status of 0 or 1, with the latter group representing 58.6% of the patients.
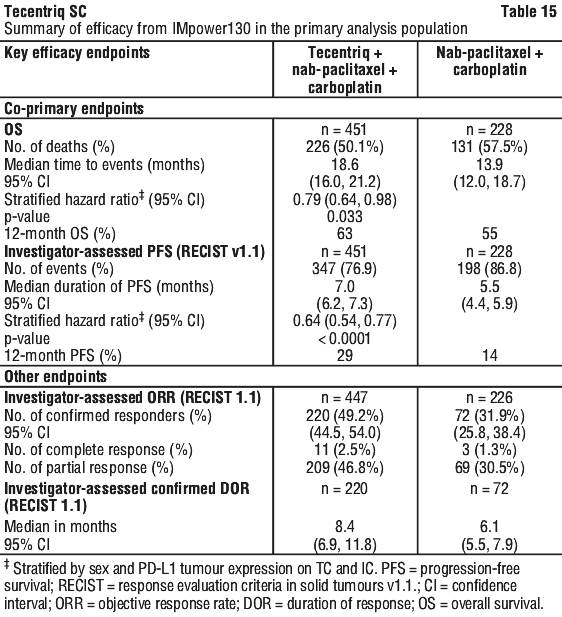
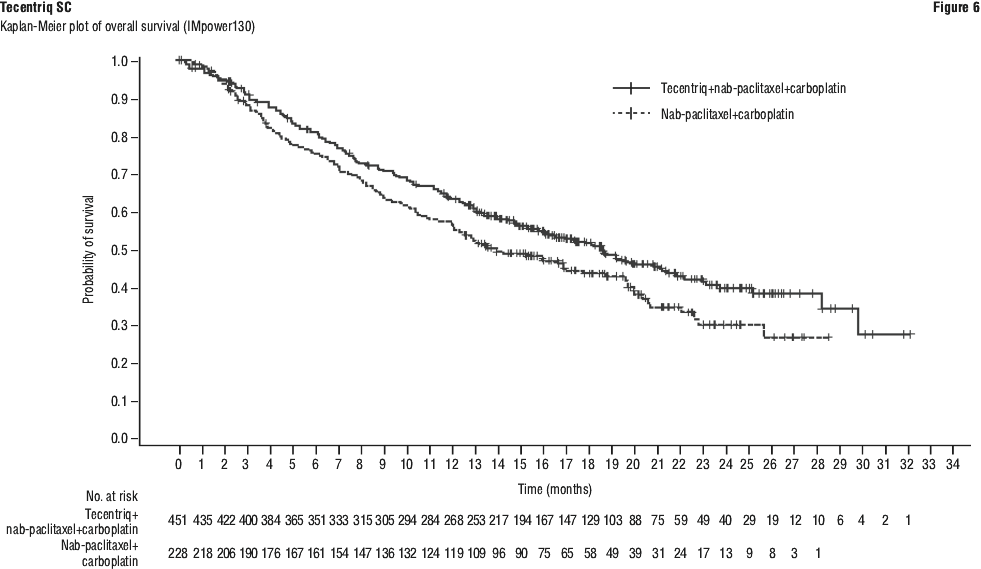
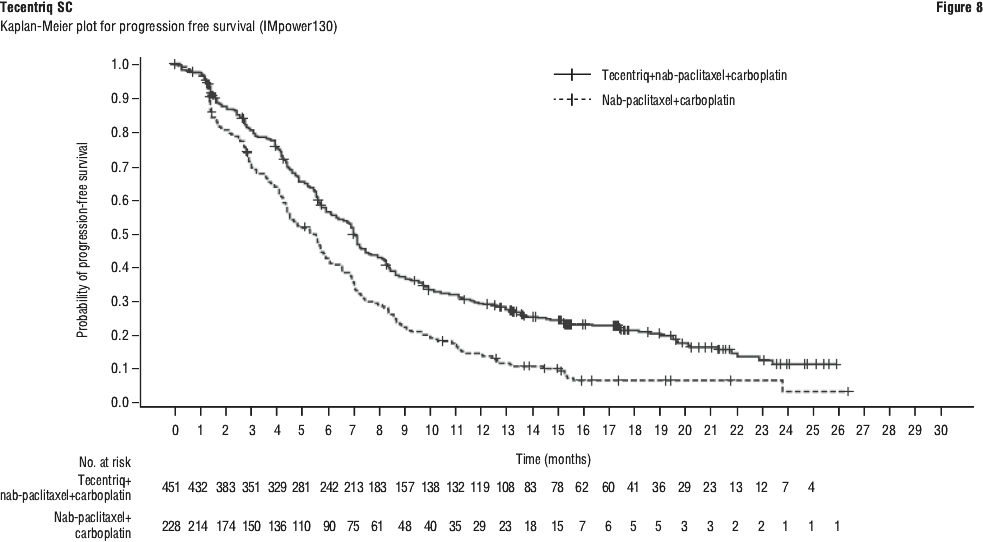
The primary analysis was conducted in all patients, excluding those with EGFR or ALK genomic tumour aberrations (n = 679). Patients had a median survival follow up time of 18.6 months. Improvements in OS and PFS were demonstrated with Tecentriq + nab-paclitaxel + carboplatin compared to the control. The key results are summarised in Table 15 and Kaplan-Meier curves for OS and PFS are presented in Figures 6 and 8, respectively.
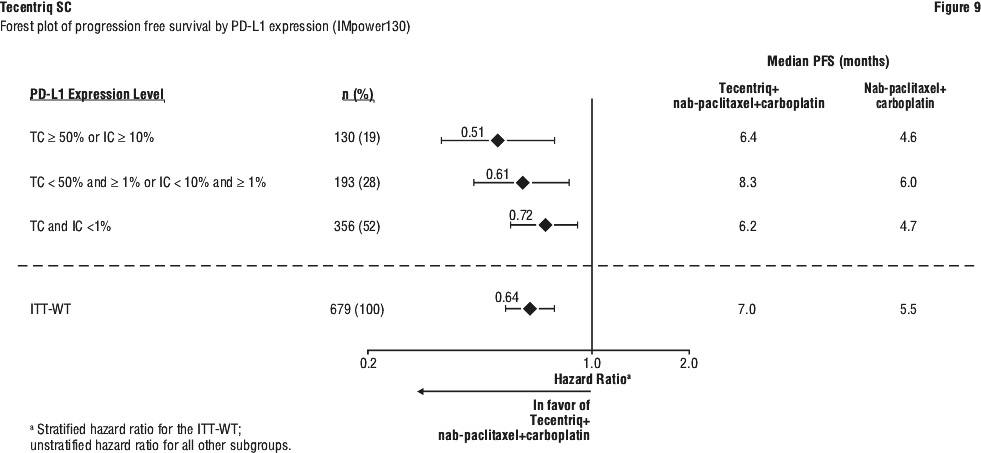
All PD-L1 subgroups, regardless of expression, derived benefit in terms of OS and PFS; the results are summarised in Figures 7 and 9. Consistent OS and PFS benefit was demonstrated in all other pre-specified subgroups, with the exception of patients with liver metastases who did not show improved OS with Tecentriq, nab-paclitaxel and carboplatin, compared to nab-paclitaxel and carboplatin (HR of 1.04, 95% CI: 0.63, 1.72).
Approximately 66% of patients in the nab-paclitaxel and carboplatin arm received any anti-cancer therapy after disease progression compared to 39% in the Tecentriq, nab-paclitaxel and carboplatin arm. These included, approximately 59% of patients in the nab-paclitaxel and carboplatin arm received any cancer immunotherapy after disease progression, which includes Tecentriq as crossover (41% of all patients), compared to 7.3% in the Tecentriq, nab-paclitaxel and carboplatin arm.

 The study also evaluated physical function and patient reported treatment-related symptoms using the EORTC QLQ-C30 and EORTC QLQ-LC13 measures. On average, patients who received Tecentriq with nab-paclitaxel and carboplatin reported high functioning and no clinically meaningful worsening in treatment-related symptoms. There was no difference in delay of lung-related symptoms (dyspnoea, cough and chest pain) however patients receiving Tecentriq, nab-paclitaxel and carboplatin reported less worsening of these symptoms over time.
The study also evaluated physical function and patient reported treatment-related symptoms using the EORTC QLQ-C30 and EORTC QLQ-LC13 measures. On average, patients who received Tecentriq with nab-paclitaxel and carboplatin reported high functioning and no clinically meaningful worsening in treatment-related symptoms. There was no difference in delay of lung-related symptoms (dyspnoea, cough and chest pain) however patients receiving Tecentriq, nab-paclitaxel and carboplatin reported less worsening of these symptoms over time.
2L NSCLC. OAK (GO28915).
A phase III, open-label, multicentre, international, randomised study, OAK (GO28915), was conducted to evaluate the efficacy and safety of Tecentriq compared with docetaxel in patients with locally advanced or metastatic NSCLC who have progressed during or following a platinum-containing regimen. A total of 1225 patients were enrolled, with the primary analysis population consisting of the first 850 randomised patients. Eligible patients were stratified by PD-L1 expression status in tumour-infiltrating immune cells (IC), by the number of prior chemotherapy regimens, and by histology. Tumour specimens were evaluated prospectively for PD-L1 expression on TCs and ICs using the VENTANA PD-L1 (SP142) Assay. Patients were randomised (1:1) to receive either Tecentriq or docetaxel. This study excluded patients who had a history of autoimmune disease, active or corticosteroid-dependent brain metastases, administration of a live, attenuated vaccine within 28 days prior to enrolment, administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to enrolment. Tumour assessments were conducted every 6 weeks for the first 36 weeks, and every 9 weeks thereafter.
The demographic and baseline disease characteristics of the primary analysis population were well balanced between the treatment arms. The median age was 64 years (range: 33 to 85), and 61% of patients were male. The majority of patients were White (70%). Approximately three-quarters of patients had non-squamous disease (74%), 10% had known EGFR mutation, 0.2% had known ALK rearrangements, 10% had CNS metastases at baseline, and most patients were current or previous smokers (82%). Baseline ECOG performance status was 0 (37%) or 1 (63%). Seventy five percent of patients received only one prior platinum-based therapeutic regimen.
Tecentriq was administered as a fixed dose of 1200 mg by IV infusion every 3 weeks. No dose reduction was allowed. Patients were treated until loss of clinical benefit as assessed by the investigator. Docetaxel was administered at 75 mg/m2 by IV infusion on day 1 of each 21 day cycle until disease progression. For all treated patients, the median duration of treatment was 2.1 months for the docetaxel arm and 3.4 months for the Tecentriq arm.
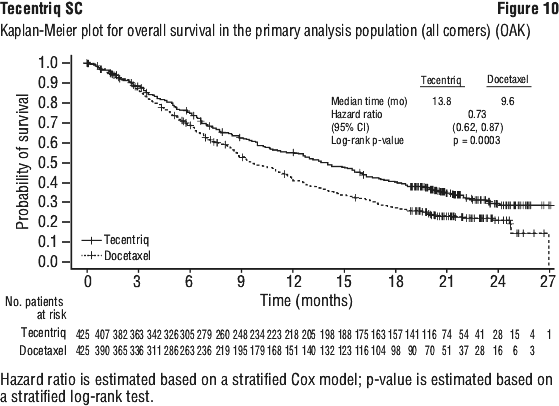
The primary efficacy endpoint was OS. The key results of this study with a median survival follow-up of 21 months are summarised in Table 16. Kaplan-Meier curves for OS in the ITT population are presented in Figure 10. Figure 11 summarises the results of OS in the ITT and PD-L1 subgroups, demonstrating OS benefit with Tecentriq in all subgroups, including those with PD-L1 expression < 1% in TC and IC.

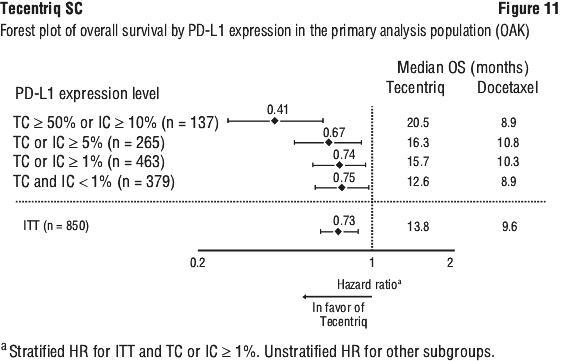 An improvement in OS was observed with Tecentriq compared to docetaxel in both non-squamous NSCLC patients (hazard ratio [HR] of 0.73, 95% CI: 0.60, 0.89; median OS of 15.6 vs. 11.2 months for Tecentriq and docetaxel, respectively) and squamous NSCLC patients (HR of 0.73, 95% CI: 0.54, 0.98; median OS of 8.9 vs. 7.7 months for Tecentriq and docetaxel, respectively). The observed OS improvement was consistently demonstrated across subgroups of patients including those with brain metastases at baseline (HR of 0.54, 95% CI: 0.31, 0.94; median OS of 20.1 vs. 11.9 months for Tecentriq and docetaxel respectively) and patients who were never smokers (HR of 0.71, 95% CI: 0.47, 1.08; median OS of 16.3 vs. 12.6 months for Tecentriq and docetaxel, respectively). However, patients with EGFR mutations did not show improved OS with Tecentriq compared to docetaxel (HR of 1.24, 95% CI: 0.71, 2.18; median OS of 10.5 vs. 16.2 months for Tecentriq and docetaxel, respectively).
An improvement in OS was observed with Tecentriq compared to docetaxel in both non-squamous NSCLC patients (hazard ratio [HR] of 0.73, 95% CI: 0.60, 0.89; median OS of 15.6 vs. 11.2 months for Tecentriq and docetaxel, respectively) and squamous NSCLC patients (HR of 0.73, 95% CI: 0.54, 0.98; median OS of 8.9 vs. 7.7 months for Tecentriq and docetaxel, respectively). The observed OS improvement was consistently demonstrated across subgroups of patients including those with brain metastases at baseline (HR of 0.54, 95% CI: 0.31, 0.94; median OS of 20.1 vs. 11.9 months for Tecentriq and docetaxel respectively) and patients who were never smokers (HR of 0.71, 95% CI: 0.47, 1.08; median OS of 16.3 vs. 12.6 months for Tecentriq and docetaxel, respectively). However, patients with EGFR mutations did not show improved OS with Tecentriq compared to docetaxel (HR of 1.24, 95% CI: 0.71, 2.18; median OS of 10.5 vs. 16.2 months for Tecentriq and docetaxel, respectively).
Based on patient responses to the EORTC QLQ-LC13 questionnaire in the OAK study, the hazard ratio (Tecentriq versus docetaxel) for time to deterioration of patient-reported pain in chest was 0.71 (95% CI: 0.49, 1.05; median not reached in either arm). The time to deterioration in other lung cancer symptoms (i.e. cough, dyspnoea, and arm/shoulder pain) was similar between arms. A delay in the time to deterioration (TTD) in physical function (HR, 0.75; 95% CI, 0.58-0.98) was observed with Tecentriq. These results should be interpreted with caution due to the open-label design of the study and lack of multiplicity control for these study endpoints.
POPLAR (GO28753).
A phase II, multicentre, international, randomised, open-label, controlled study, POPLAR (GO28753) was conducted in patients with locally advanced or metastatic NSCLC who progressed during or following a platinum containing regimen, regardless of PD-L1 expression. The primary efficacy outcome was overall survival. A total of 287 patients were randomised 1:1 to receive either Tecentriq (1200 mg by intravenous infusion every 3 weeks until loss of clinical benefit) or docetaxel (75 mg/m2 by intravenous infusion on day 1 of each 3 week cycle until disease progression). Randomisation was stratified by PD-L1 expression status on IC (according to the VENTANA PD-L1 (SP142) Assay), by the number of prior chemotherapy regimens and by histology. An updated analysis with a total of 200 deaths observed and a median survival follow up of 22 months showed a median OS of 12.6 months in patients treated with Tecentriq, vs. 9.7 months in patients treated with docetaxel (HR of 0.69, 95% CI: 0.52, 0.92). ORR was 15.3% vs. 14.7% and median DOR was 18.6 months vs. 7.2 months for Tecentriq vs. docetaxel, respectively.
Intravenous Tecentriq.
Small cell lung cancer. IMpower133 (GO30081).
A Phase I/III, randomised, multicentre, double-blind, placebo controlled study, IMpower133 (GO30081), was conducted to evaluate the efficacy and safety of Tecentriq in combination with carboplatin and etoposide in patients with chemotherapy-naïve ES-SCLC. A total of 403 patients were randomised (1:1) to receive one of the treatment regimens described in Table 17. Randomisation was stratified by sex, ECOG performance status, and presence of brain metastases.
This study excluded patients who had active or untreated CNS metastases; history of autoimmune disease; administration of live, attenuated vaccine within 4 weeks prior to randomisation; administration of systemic immunosuppressive medications within 1 week prior to randomisation. Tumour assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter. Patients treated beyond disease progression had tumour assessment conducted every 6 weeks until treatment discontinuation.
 The demographic and baseline disease characteristics of the primary analysis population were well balanced between the treatment arms. The median age was 64 years (range: 26 to 90 years). The majority of patients were male (65%), White (80%), and 9% had brain metastases and most patients were current or previous smokers (97%). Baseline ECOG performance status was 0 (35%) or 1 (65%).
The demographic and baseline disease characteristics of the primary analysis population were well balanced between the treatment arms. The median age was 64 years (range: 26 to 90 years). The majority of patients were male (65%), White (80%), and 9% had brain metastases and most patients were current or previous smokers (97%). Baseline ECOG performance status was 0 (35%) or 1 (65%).
At the time of the primary analysis, patients had a median survival follow up time of 13.9 months. The key results are summarised in Table 18. Kaplan-Meier curves for OS and PFS are presented in Figures 12 and 13.
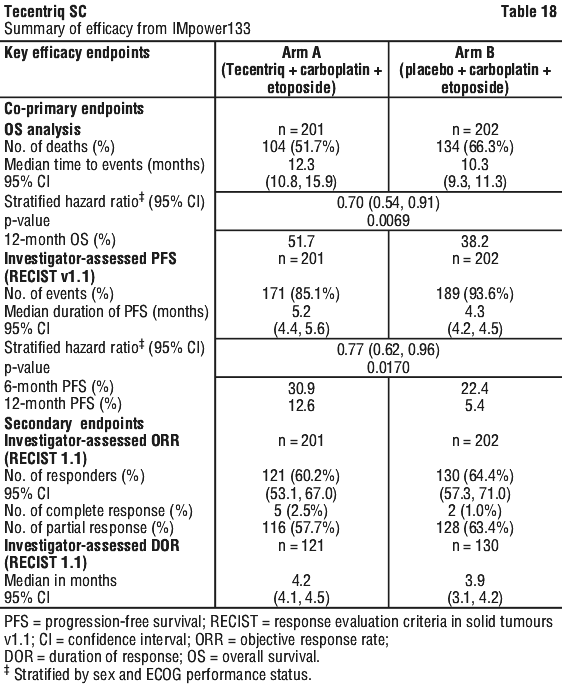
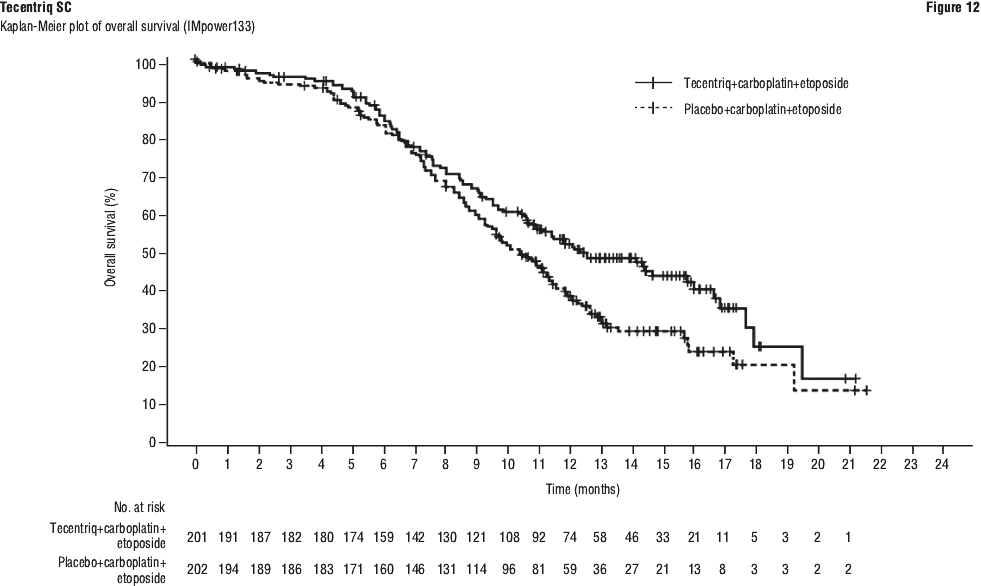
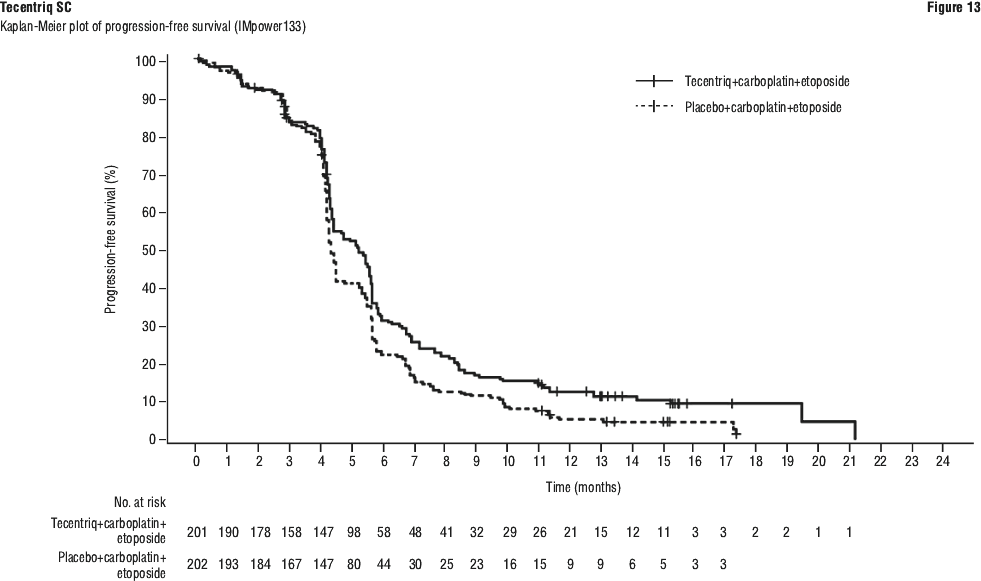
Intravenous Tecentriq.
Urothelial carcinoma. IMvigor210 (GO29293).
The efficacy of Tecentriq was investigated in IMvigor210 (Cohort 1) (GO29293), a multicentre, open-label, single-arm trial that included 119 patients with locally advanced or metastatic urothelial carcinoma who were ineligible for cisplatin-containing chemotherapy and were either previously untreated or had disease progression at least 12 months after neoadjuvant or adjuvant chemotherapy. Patients were considered cisplatin-ineligible if they met any one of the following criteria at study entry: impaired renal function [creatinine clearance (CLcr) of 30 to 59 mL/min], ECOG performance status (PS) of 2, hearing loss of ≥ 25 decibels (dB) at two contiguous frequencies, or Grades 2 to 4 peripheral neuropathy. This study excluded patients who had: a history of autoimmune disease; active or corticosteroid-dependent brain metastases; administration of a live, attenuated vaccine within 28 days prior to enrolment; or administration of systemic immunostimulatory agents within 6 weeks or systemic immunosuppressive medications within 2 weeks prior to enrolment.
Patients received Tecentriq 1200 mg as an intravenous infusion every 3 weeks until unacceptable toxicity or disease progression. Tumour response assessments were conducted every 9 weeks for the first 54 weeks and every 12 weeks thereafter. Major efficacy outcome measures included confirmed overall response rate (ORR) as assessed by independent review facility (IRF) using Response Evaluation Criteria in Solid Tumours (RECIST v1.1), duration of response (DoR) and overall survival (OS).
In this study, the median age was 73 years, 81% were male, and 91% were White. Thirty-five percent of patients had non-bladder urothelial carcinoma and 66% had visceral metastases. Eighty percent of patients had an ECOG PS of 0 or 1. Reasons for ineligibility for cisplatin containing chemotherapy were: 70% had impaired renal function, 20% had an ECOG PS of 2, 14% had a hearing loss of ≥ 25 dB, and 6% had Grades 2 to 4 peripheral neuropathy at baseline. Twenty percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy.
Tumour specimens were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory, and the results were used to define subgroups for pre-specified analyses. Of the 119 patients, 27% were classified as having PD-L1 expression of ≥ 5% (defined as PD-L1 stained tumour-infiltrating immune cells [IC] covering ≥ 5% of the tumour area). The remaining 73% of patients were classified as having PD-L1 expression of < 5% (PD-L1 stained tumour infiltrating IC covering < 5% of the tumour area).
Among the 32 patients with PD-L1 expression of ≥ 5%, median age was 67 years, 81% were male, 19% female, and 88% were White. Twenty-eight percent of patients had non-bladder urothelial carcinoma and 56% had visceral metastases. Seventy-two percent of patients had an ECOG PS of 0 or 1. Reasons for ineligibility for cisplatin-containing chemotherapy were: 66% had impaired renal function, 28% had an ECOG PS of 2, 16% had a hearing loss ≥ 25 dB, and 9% had Grades 2 - 4 peripheral neuropathy at baseline. Thirty-one percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy.
Confirmed ORR in all patients and the two PD-L1 subgroups are summarised in Table 19. The median follow-up time for this study was 14.4 months. In 24 patients with disease progression following neoadjuvant or adjuvant therapy, the ORR was 33% (95% CI: 16%, 55%).
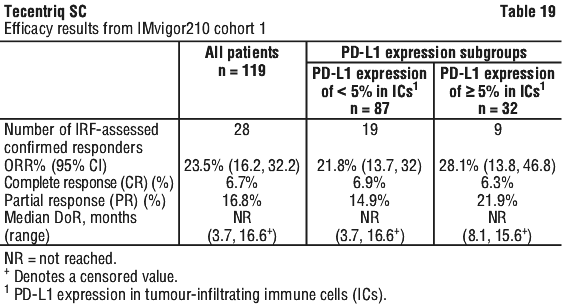
IMvigor130 (WO30070).
The phase III, multicentre, randomised study IMvigor130 (WO30070) enrolled patients with locally advanced or metastatic urothelial carcinoma who had not received prior systemic therapy in the metastatic setting and were eligible for platinum-containing chemotherapy. Cisplatin eligibility was by investigator judgement, guided by Galsky criteria. Tumour specimens were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory. A total of 1213 patients were enrolled and were randomised 1:1:1 to receive Tecentriq, chemotherapy or both, as summarised in Table 20. Changes to study design over time resulted in uneven arm sizes and a population enriched for patients who were cisplatin ineligible. Randomisation was stratified by PD-L1 tumour expression on tumour-infiltrating immune cells (IC), Bajorin model risk factor score/ liver metastasis and whether the pre-randomisation investigator's choice of chemotherapy was cisplatin or carboplatin.
At an unplanned early analysis, based on an independent Data Monitoring Committee (iDMC) recommendation, accrual of patients on the Tecentriq monotherapy treatment arm whose tumours had a low PD-L1 expression (PD-L1 stained tumour-infiltrating immune cells [IC] covering < 5% of the tumour area) was stopped. This was recommended due to an observation of decreased overall survival (OS) for this subgroup at an unplanned early analysis, which occurred after the vast majority of patients had already been enrolled. No other changes were recommended for the study, including any change of therapy for patients who had already been randomised to and were receiving treatment in the monotherapy arm.
 The co-primary efficacy endpoints for IMvigor130 were investigator-assessed progression-free survival (PFS) and overall survival (OS). Secondary efficacy endpoints were objective response rate (ORR) and duration of response (DOR). The median survival follow up was 13.4 months (range: 0.0 - 71.7 months).
The co-primary efficacy endpoints for IMvigor130 were investigator-assessed progression-free survival (PFS) and overall survival (OS). Secondary efficacy endpoints were objective response rate (ORR) and duration of response (DOR). The median survival follow up was 13.4 months (range: 0.0 - 71.7 months).
The study did not meet the co-primary endpoint of OS and did not demonstrate a comparative clinical benefit of atezolizumab over platinum-based chemotherapy, whether given as monotherapy or in combination with chemotherapy, in the intention-to-treat (ITT) population. A description of selected exploratory findings from IMvigor130 are provided in Table 21, including amongst the stratum of patients for whom the investigator's pre-randomisation choice of platinum agent was carboplatin, and in subgroups based on whether PD-L1 stained tumour-infiltrating immune cells [IC] covered < 5% (PD-L1 low) or at least 5% higher (PD-L1 high) of the tumour area.
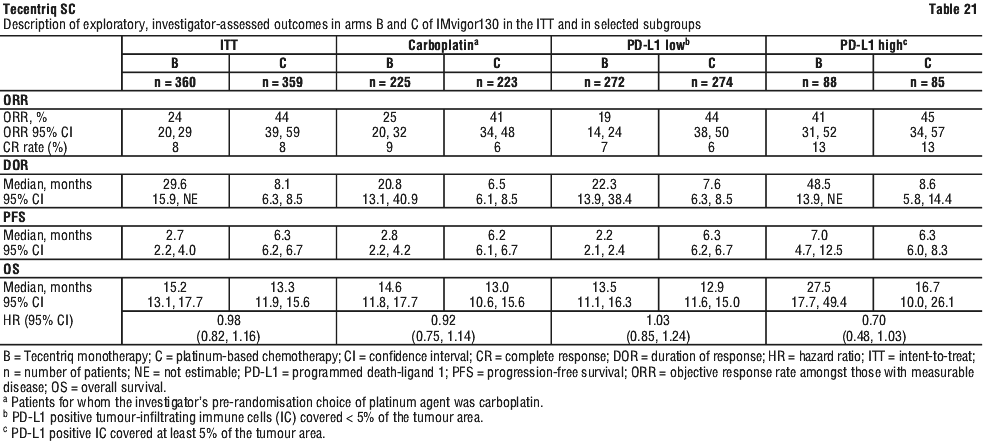
Intravenous Tecentriq.
Hepatocellular carcinoma. IMbrave150 (YO40245).
A global phase III, randomised, multi-centre, open-label study, IMbrave150 (YO40245), was conducted to evaluate the efficacy and safety of Tecentriq in combination with bevacizumab, in patients with locally advanced or metastatic and/or unresectable HCC, who have not received prior systemic treatment. A total of 501 patients were randomised (2:1) to receive either Tecentriq 1200 mg and 15 mg/kg of bevacizumab every 3 weeks administered via IV infusion, or sorafenib 400 mg orally twice per day. Randomisation was stratified by geographic region (Asia excluding Japan vs. rest of world), macrovascular invasion and/or extrahepatic spread (presence vs. absence), baseline AFP (< 400 vs. ≥ 400 nanogram/mL) and ECOG performance status (0 vs. 1). Patients in both arms received treatment until loss of clinical benefit, or unacceptable toxicity. Patients could discontinue either Tecentriq or bevacizumab (e.g. due to adverse events) and continue on single-agent therapy until loss of clinical benefit or unacceptable toxicity associated with the single-agent.
The study enrolled adults who were ECOG 0/1. Patients were required to be evaluated for the presence of varices within 6 months prior to treatment, and were excluded if they had variceal bleeding within 6 months prior to treatment, untreated or incompletely treated varices with bleeding or high risk of bleeding. Patients were also excluded if they had Child-Pugh B or C cirrhosis; moderate or severe ascites; history of hepatic encephalopathy; a history of autoimmune disease; administration of a live, attenuated vaccine within 4 weeks prior to randomisation; administration of systemic immunostimulatory agents within 4 weeks, or systemic immunosuppressive medications within 2 weeks prior to randomisation; untreated or corticosteroid-dependent brain metastases. Tumour assessments were performed every 6 weeks for the first 54 weeks and every 9 weeks thereafter.
The demographic and baseline disease characteristics of the study population were balanced between the treatment arms. The median age was 65 years (range: 26 to 88 years) and 83% were male. The majority of patients were Asian (57%) and White (35%); 40% were from Asia (excluding Japan). Approximately 75% of patients presented with macrovascular invasion and/or extrahepatic spread and 37% had a baseline AFP ≥ 400 nanogram/mL. Baseline ECOG performance status was 0 (62%) or 1 (38%). HCC risk factors were Hepatitis B virus infection in 48% of patients, Hepatitis C virus infection in 22% of patients, and non-viral disease in 31% of patients. HCC was categorised as Barcelona Clinic Liver Cancer (BCLC) stage C in 82% of patients, stage B in 16% of patients, and stage A in 3% of patients.
The co-primary efficacy endpoints were overall survival (OS) and independent review facility (IRF)-assessed progression free survival (PFS) according to RECIST v1.1. Additional efficacy outcome measures were IRF-assessed objective response rate (ORR) per RECIST v1.1 and HCC modified RECIST (mRECIST). Efficacy results are summarised in Table 22. Kaplan-Meier curves for OS and PFS are presented in Figures 14 and 15.

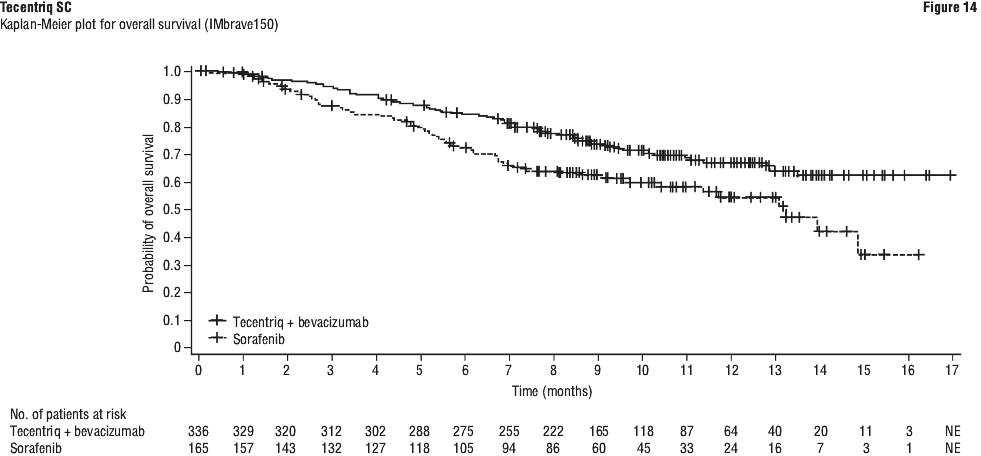
 The study also evaluated patient-reported outcomes using the EORTC QLQ-C30 and EORTC QLQ-HCC18 questionnaires. Treatment with Tecentriq and bevacizumab delayed clinically meaningful deterioration of patient-reported physical functioning, role functioning, global health status/quality of life and key symptoms (i.e. appetite loss, diarrhoea, fatigue and pain) versus sorafenib.
The study also evaluated patient-reported outcomes using the EORTC QLQ-C30 and EORTC QLQ-HCC18 questionnaires. Treatment with Tecentriq and bevacizumab delayed clinically meaningful deterioration of patient-reported physical functioning, role functioning, global health status/quality of life and key symptoms (i.e. appetite loss, diarrhoea, fatigue and pain) versus sorafenib.
Immunogenicity.
Tecentriq SC. BP40657 (IMscin001).
Exploratory analyses showed that the subset of patients in the Tecentriq SC arm who were treatment-emergent ADA positive (19%) appeared to have similar efficacy (effect on progression-free survival) as compared to patients who tested negative for treatment-emergent ADA (81%). Median time to progression was 2.8 months (95% CI: 1.4, 4.2) in the treatment-emergent ADA positive patients and 2.9 months (95% CI: 2.7, 4.2) in the treatment-emergent ADA negative patients.
Intravenous Tecentriq. GO29436 (IMpower150).
Exploratory analyses adjusting for imbalances in baseline health and disease characteristics showed that the subset of patients in the four drug regimen arm who were ADA positive by week 4 (32%) appeared to have similar efficacy (effect on overall survival) as compared to patients who tested negative for treatment-emergent ADA by week 4 (68%). The OS hazard ratio comparing the ADA-positive subgroup of the four drug regimen to the three drug regimen (control) was 0.77 (95% CI: 0.57, 1.02). The OS hazard ratio comparing the ADA-negative subgroup to control was 0.73 (95% CI: 0.58, 0.93).
GO28915 (OAK).
Exploratory analyses adjusting for imbalances in baseline health and disease characteristics showed that the subset of patients who were ADA positive by week 4 (22%) appeared to have similar efficacy (effect on overall survival) as compared to patients who tested negative for treatment-emergent ADA by week 4 (78%). The OS hazard ratio comparing the ADA-positive subgroup of the Tecentriq arm to docetaxel was 0.75 (95% CI: 0.57, 0.98). The OS hazard ratio comparing the ADA-negative subgroup to docetaxel was 0.69 (95% CI: 0.56, 0.84).
YO40245 (IMbrave150).
Exploratory analyses adjusting for imbalances in baseline health and disease characteristics showed that the subset of patients (20%) who were ADA-positive by week 6 appeared to have reduced efficacy (effect on OS) as compared to patients (80%) who tested negative for treatment-emergent ADA by week 6. ADA-positive patients by week 6 appeared to have similar overall survival compared to sorafenib-treated patients. The OS hazard ratio comparing the ADA-positive subgroup of the Tecentriq and bevacizumab arm to sorafenib was 0.95 (95% CI: 0.57, 1.59). The OS hazard ratio comparing the ADA-negative subgroup to sorafenib was 0.41 (95% CI: 0.27, 0.62).
5.2 Pharmacokinetic Properties
Tecentriq SC.
Atezolizumab model-predicted exposure metrics following Tecentriq SC (1875 mg SC administered every 3 weeks) and intravenous atezolizumab (1200 mg IV administered every 3 weeks) administration in the IMscin001 study are shown in Table 23. Atezolizumab Cycle 1 observed serum Ctrough (i.e. pre-dose cycle 2) showed non-inferiority of atezolizumab within Tecentriq SC to intravenous atezolizumab, with a geometric mean ratio (GMR) of 1.05 (90% CI: 0.88 - 1.24).
The GMR for Cycle 1 model-predicted for AUC from 0 to 21 days (AUC0-21d) was 0.87 (90% CI: 0.83 - 0.92). The maximum systemic accumulation ratio following 1875 mg of Tecentriq SC administered every 3 weeks is 2.2. The model-predicted Ctrough and AUC at steady state were comparable for Tecentriq SC and intravenous atezolizumab (see Table 23).

Absorption.
Tecentriq SC.
Based on population PK analysis, the absolute bioavailability was 72% and the first-order absorption rate (Ka) is 0.3 (1/day). The atezolizumab geometric mean maximum serum concentration (Cmax) was 189 mcg/mL and median time to maximum serum concentration (Tmax) was 4.5 days (median; 2.2-9.0 days min-max).
Distribution.
A population pharmacokinetic analysis indicates that central compartment volume of distribution (V1) is 3.28 L and volume at steady-state (Vss) is 6.91 L in the typical patient.
Metabolism.
The metabolism of atezolizumab has not been directly studied. Antibodies are cleared principally by catabolism.
Excretion.
A population pharmacokinetic analysis indicates that the clearance of atezolizumab is 0.200 L/day and the typical terminal elimination half-life (t1/2) is 27 days.
Pharmacokinetics in special populations.
Hepatic impairment.
No dedicated studies of atezolizumab have been conducted in patients with hepatic impairment. In the population pharmacokinetic analysis, there were no clinically important differences in the clearance of intravenous or subcutaneously administered atezolizumab between patients with mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1.0 to 1.5 x ULN and any AST) or moderate hepatic impairment (bilirubin > 1.5 to 3 x ULN and any AST). No data are available in patients with severe (bilirubin > 3.0 x ULN and any AST) hepatic impairment. Hepatic impairment was defined by the National Cancer Institute (NCI) criteria of hepatic dysfunction (see Section 4.2 Dose and Method of Administration). The effect of moderate or severe hepatic impairment (bilirubin > 1.5 x to 3 x ULN and any AST or bilirubin > 3 x ULN and any AST) on the pharmacokinetics of atezolizumab is unknown.
Renal impairment.
No dedicated studies of atezolizumab have been conducted in patients with renal impairment. In the population pharmacokinetic analysis, no clinically important differences in the clearance of intravenous atezolizumab were found in patients with mild (eGFR 60 to 89 mL/min/1.73 m2; n = 208) or moderate (eGFR 30 to 59 mL/min/1.73 m2; n = 116) renal impairment compared to patients with normal (eGFR greater than or equal to 90 mL/min/1.73 m2; n = 140) renal function. Only a few patients had severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2; n = 8) (see Section 4.2 Dose and Method of Administration). The effect of severe renal impairment on the pharmacokinetics of atezolizumab is unknown.
No clinically relevant differences in the clearance of subcutaneous atezolizumab were found in patients with mild (eGFR 60 to 89 mL/min/1.73 m2; n = 111) or moderate (eGFR 30 to 59 mL/min/1.73 m2; n = 32) renal impairment compared to patients with normal (eGFR greater than or equal to 90 mL/min/1.73 m2; n = 103) renal function.
Elderly.
No dedicated studies of atezolizumab have been conducted in elderly patients. The effect of age on the pharmacokinetics of atezolizumab was assessed in a population pharmacokinetic analysis. Age was not identified as a significant covariate influencing intravenous atezolizumab pharmacokinetics based on patients of age range of 21 - 89 years (n = 472), and median of 62 years of age. No clinically important difference was observed in the pharmacokinetics of atezolizumab among patients < 65 years (n = 274), patients between 65 - 75 years (n = 152) and patients > 75 years (n = 46) (see Section 4.2 Dose and Method of Administration).
No clinically relevant difference was observed in the pharmacokinetics of subcutaneous atezolizumab among patients < 65 years (n = 138), patients between 65 - 75 years (n = 89), and patients > 75 years of age (n = 19).
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been conducted with atezolizumab.
Carcinogenicity.
No carcinogenicity studies have been conducted with atezolizumab.
Subcutaneous formulation.
Tecentriq SC contains vorhyaluronidase alfa (see Section 6.1 List of Excipients). No carcinogenicity or genotoxicity studies were conducted for vorhyaluronidase alfa.
6 Pharmaceutical Particulars
6.1 List of Excipients
Vorhyaluronidase alfa (an enzyme used to increase the dispersion and absorption of co-administered drugs when administered subcutaneously), histidine, acetic acid, methionine, sucrose, polysorbate 20 and water for injections.
6.2 Incompatibilities
No incompatibilities have been observed between Tecentriq SC and polyvinyl chloride (PVC), polypropylene (PP), polycarbonate (PC), stainless steel (ss), and polyurethanes (PU).
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
From a microbiological point of view, the product should be used immediately once transferred from the vial to the syringe since the medicine does not contain any antimicrobial-preservative.
The closed syringe can be stored at ≤ 25°C for up to 8 hours in diffuse daylight and in the refrigerator (2°C to 8°C) for up to 72 hours (3 days).
6.4 Special Precautions for Storage
Store the vials at 2°C to 8°C. Do not freeze.
Tecentriq SC vials should be kept in the outer carton in order to protect from light. Do not shake.
This medicine should not be used after the expiry date (EXP) shown on the pack.
6.5 Nature and Contents of Container
Tecentriq SC is available in a single-use glass vial containing 15 mL solution in a pack size of 1 vial.
6.6 Special Precautions for Disposal
The release of medicines into the environment should be minimised. Medicines should not be disposed of via wastewater and disposal through household waste should be avoided. Unused or expired medicine should be returned to a pharmacy for disposal.
6.7 Physicochemical Properties
Chemical structure.
Atezolizumab is an engineered, humanised, monoclonal antibody that directly binds to PD-L1 and blocks interactions with the PD-1 and B7.1 receptors. Atezolizumab is a non-glycosylated IgG1 immunoglobulin that has a calculated molecular mass of 145 kDa.
CAS number.
1380723-44-3.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
