1 Name of Medicine
Teclistamab.
2 Qualitative and Quantitative Composition
Tecvayli (teclistamab) is a humanised immunoglobulin G4-proline, alanine, alanine (IgG4-PAA) bispecific antibody targeting the B cell maturation antigen (BCMA) and CD3 receptors, produced in a mammalian cell line (Chinese hamster ovary [CHO]) using recombinant DNA technology.
Tecvayli is available in the following presentations:
Each 3 mL vial contains 30 mg of teclistamab (10 mg of teclistamab per mL).
Each 1.7 mL vial contains 153 mg of teclistamab (90 mg of teclistamab per mL).
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Tecvayli is a colourless to light yellow preservative-free solution for injection.
4.1 Therapeutic Indications
Tecvayli as monotherapy has provisional approval in Australia and is indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least three prior therapies, including a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody.
The decision to approve this indication has been made on the basis of the overall response rate in a single arm study. Continued approval of this indication depends on verification and description of benefit in confirmatory trials.
4.2 Dose and Method of Administration
Treatment with Tecvayli should be initiated and supervised by physicians experienced in the treatment of multiple myeloma.
Dosage - adults (18 years of age and older).
Tecvayli should be administered by subcutaneous injection only.
Administer pretreatment medications prior to each dose of the Tecvayli step-up dosing schedule (see Pretreatment medications).
Recommended dosing schedule.
The recommended dosing schedule for Tecvayli is provided in Table 1. The recommended dosage of Tecvayli is step-up doses of 0.06 mg/kg and 0.3 mg/kg followed by 1.5 mg/kg once weekly until disease progression or unacceptable toxicity.
In patients who have a complete response or better for a minimum of 6 months, a reduced dosing frequency of 1.5 mg/kg every two weeks until disease progression or unacceptable toxicity may be considered (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Administer Tecvayli according to the step-up dosing schedule in Table 1 to reduce the incidence and severity of cytokine release syndrome (CRS). Due to the risk of cytokine release syndrome, instruct patients to remain within proximity of a healthcare facility and monitor patients for signs and symptoms daily for 48 hours after administration of all doses within the Tecvayli step-up dosing schedule (see Administration; see Section 4.4 Special Warnings and Precautions for Use, Cytokine release syndrome (CRS)).
Failure to follow the recommended doses or dosing schedule for initiation of therapy or re-initiation of therapy after dose delays may result in increased frequency and severity of adverse events related to mechanism of action, particularly cytokine release syndrome (see Dosage modifications; see Section 4.4 Special Warnings and Precautions for Use, Cytokine release syndrome (CRS)).
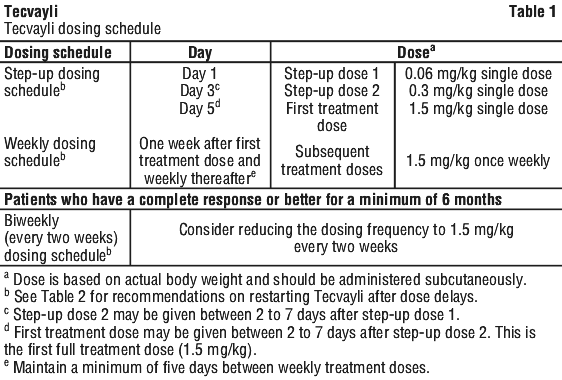 See Tables 6, 7 and 8 to determine the dosage based on predetermined weight ranges (see Administration, Preparation of Tecvayli).
See Tables 6, 7 and 8 to determine the dosage based on predetermined weight ranges (see Administration, Preparation of Tecvayli).
For guidance regarding restarting therapy with Tecvayli after dose delays, see Restarting Tecvayli after dose delays.
Pretreatment medications.
Administer the following pretreatment medications 1 to 3 hours before each dose of the Tecvayli step-up dosing schedule to reduce the risk of cytokine release syndrome (see Section 4.4 Special Warnings and Precautions for Use, Cytokine release syndrome (CRS); Section 4.8 Adverse Effects (Undesirable Effects)).
Corticosteroid (oral or intravenous dexamethasone, 16 mg or equivalent).
Antihistamine (oral or intravenous diphenhydramine, 50 mg or equivalent).
Antipyretics (oral paracetamol 500 mg to 1000 mg).
Administration of pretreatment medications may be required prior to administration of subsequent doses of Tecvayli in the following patients: (see Dosage modifications).
Patients who repeat doses within the Tecvayli step-up dosing schedule following a dose delay (see Restarting Tecvayli after dose delays).
Patients who experienced CRS following the prior dose of Tecvayli (see Dosage modifications).
Prophylaxis for herpes zoster virus reactivation.
Prior to starting treatment with Tecvayli, anti-viral prophylaxis should be considered for the prevention of herpes zoster virus reactivation per local institutional guidelines.
Restarting Tecvayli after dose delays.
If a dose of Tecvayli is delayed, restart therapy based on the recommendations listed in Table 2 and resume the treatment schedule accordingly (see Dosage - adults (18 years of age and older)). Administer pretreatment medications as indicated in Table 2 and monitor patients following administration of Tecvayli accordingly (see Pretreatment medications and Administration).
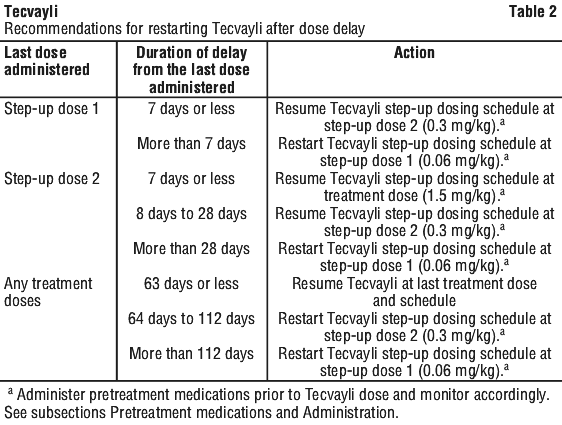
Dosage modifications.
Do not skip step-up doses of Tecvayli.
Dose reductions of Tecvayli are not recommended.
Dose delays may be required to manage toxicities related to Tecvayli (see Section 4.4 Special Warnings and Precautions for Use).
See Table 3 for recommended actions for adverse reactions following administration of Tecvayli.
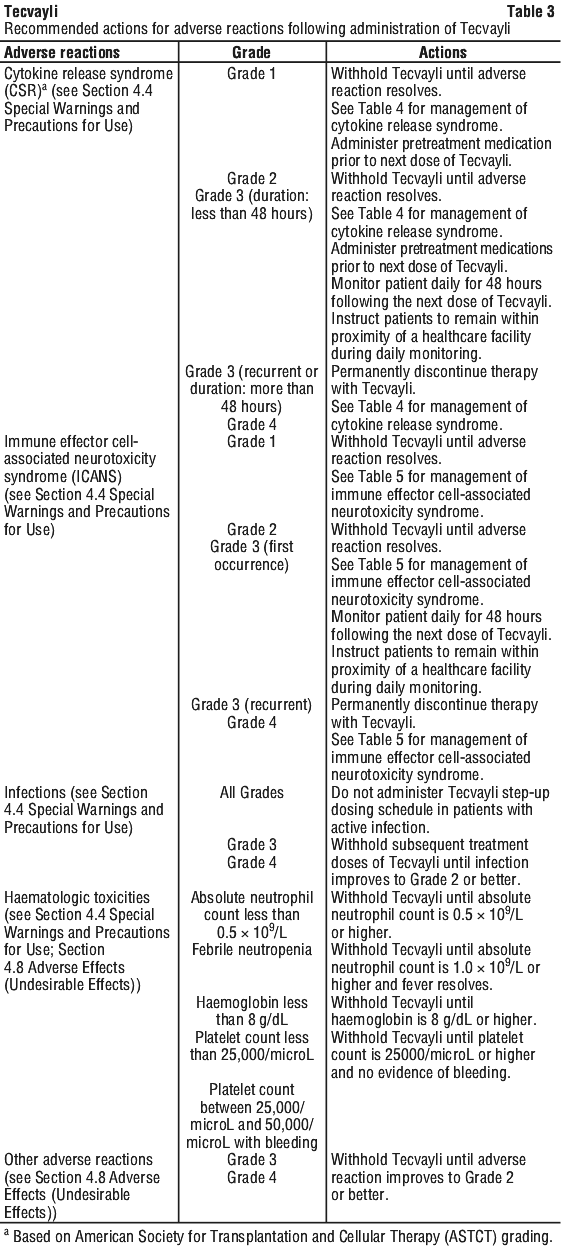
Management of severe adverse reactions.
Cytokine release syndrome (CRS).
CRS, including life-threatening or fatal reactions, may occur in patients receiving Tecvayli.
Identify CRS based on clinical presentation (see Section 4.4 Special Warnings and Precautions for Use, Cytokine release syndrome (CRS)). Evaluate and treat other causes of fever, hypoxia, and hypotension.
If CRS is suspected, withhold Tecvayli until the adverse reaction resolves (see Table 3) and manage according to the recommendations in Table 4. Administer supportive care for CRS (including but not limited to anti-pyretic agents, intravenous fluid support, vasopressors, supplemental oxygen, etc.) as appropriate. Consider laboratory testing to monitor for disseminated intravascular coagulation (DIC), haematology parameters, as well as pulmonary, cardiac, renal, and hepatic function.
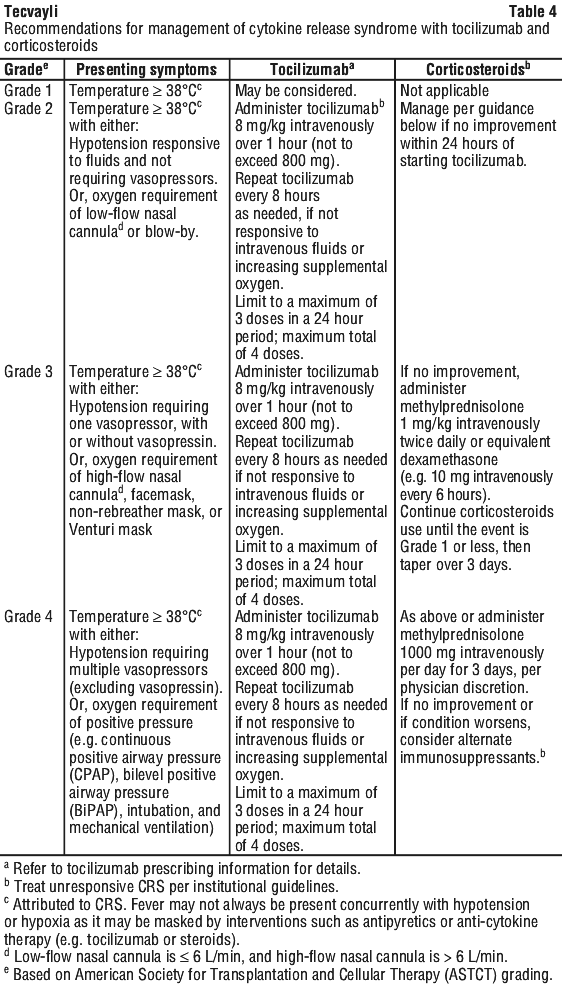
Neurologic toxicities.
Serious or life-threatening neurological toxicities, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) with or without concurrent CRS, may follow treatment with Tecvayli.
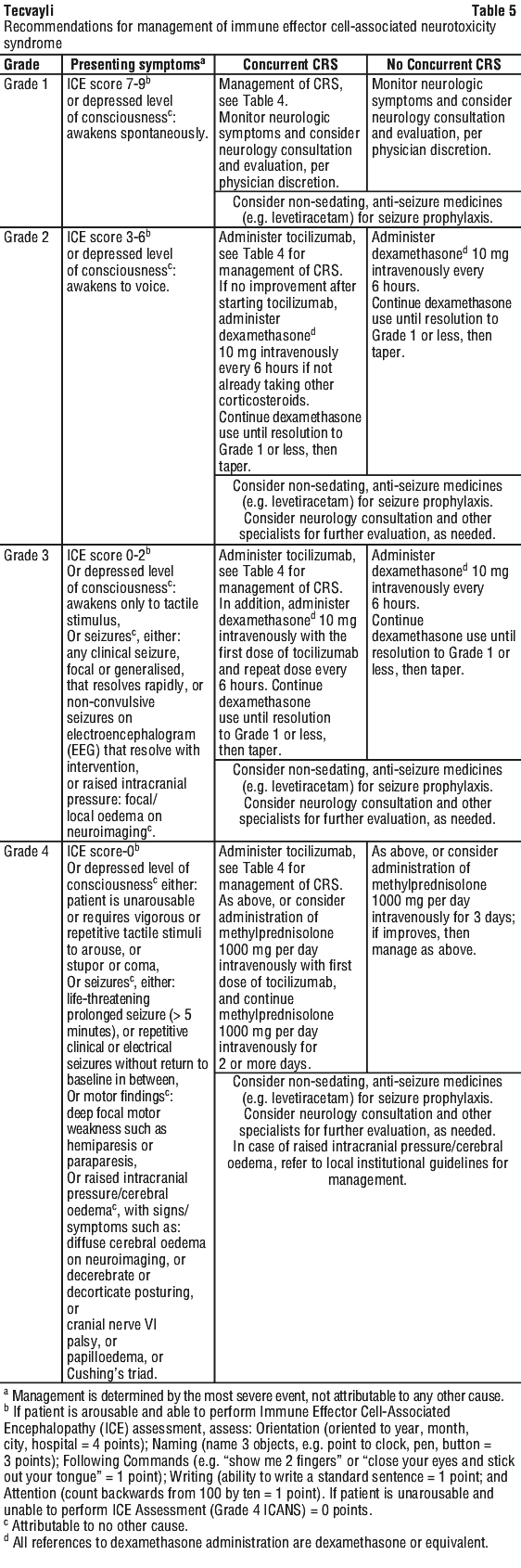
General management for neurologic toxicity (e.g. Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS) with or without concurrent CRS) is summarised in Table 5.
At the first sign of neurologic toxicity including ICANS, consider neurology evaluation. Rule out other causes of neurologic symptoms. Provide intensive care and supportive therapy for severe or life-threatening neurologic toxicities (see Section 4.4 Special Warnings and Precautions for Use, Neurologic toxicities). Withhold Tecvayli as indicated in Table 3.
Use in hepatic impairment.
No formal studies of Tecvayli in patients with hepatic impairment have been conducted.
Based on population pharmacokinetic analyses, no dose adjustment is recommended for patients with mild hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
No formal studies of Tecvayli in patients with renal impairment have been conducted.
Based on population pharmacokinetic analyses, no dose adjustment is recommended for patients with mild or moderate renal impairment (see Section 5.2 Pharmacokinetic Properties).
Administration.
Strictly follow the preparation and administration instructions provided in this section to minimise potential dosing errors with Tecvayli 10 mg/mL vial and Tecvayli 90 mg/mL vial.
Tecvayli should be administered via subcutaneous injection only. Do not administer Tecvayli intravenously.
Tecvayli should be administered by a healthcare professional with adequately trained medical personnel and appropriate medical equipment to manage severe reactions, including cytokine release syndrome (see Section 4.4 Special Warnings and Precautions for Use, Cytokine release syndrome (CRS)).
Tecvayli 10 mg/mL vial and Tecvayli 90 mg/mL vial are supplied as ready-to-use solution for injection that do not need dilution prior to administration.
Tecvayli vials of different concentrations should not be combined to achieve treatment dose.
Use aseptic technique to prepare and administer Tecvayli.
Preparation of Tecvayli.
Verify the prescribed dose for each Tecvayli injection. To minimise errors, use the following tables to prepare Tecvayli injection.
Use Table 6 to determine total dose, injection volume and number of vials required based on patient's actual body weight for step-up dose 1 using Tecvayli 10 mg/mL.
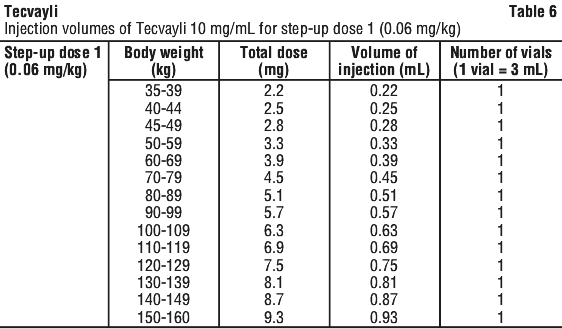 Use Table 7 to determine total dose, injection volume and number of vials required based on patient's actual body weight for step-up dose 2 using Tecvayli 10 mg/mL.
Use Table 7 to determine total dose, injection volume and number of vials required based on patient's actual body weight for step-up dose 2 using Tecvayli 10 mg/mL.
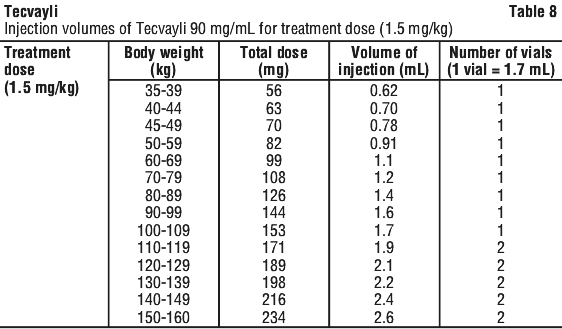
 Use Table 8 to determine total dose, injection volume and number of vials required based on patient's actual body weight for the treatment dose using Tecvayli 90 mg/mL.
Use Table 8 to determine total dose, injection volume and number of vials required based on patient's actual body weight for the treatment dose using Tecvayli 90 mg/mL.
 Remove the appropriate strength Tecvayli vial from refrigerated storage 2°C-8°C and equilibrate to ambient temperature 15°C-30°C for at least 15 minutes prior to administration. Do not warm Tecvayli in any other way.
Remove the appropriate strength Tecvayli vial from refrigerated storage 2°C-8°C and equilibrate to ambient temperature 15°C-30°C for at least 15 minutes prior to administration. Do not warm Tecvayli in any other way.
Once equilibrated, gently swirl the vial for approximately 10 seconds to mix. Do not shake.
Withdraw the required injection volume of Tecvayli from the vial(s) into an appropriately sized syringe using a transfer needle.
Each injection volume should not exceed 2.0 mL. Divide doses requiring greater than 2.0 mL equally into multiple syringes.
Tecvayli is compatible with stainless steel injection needles and polypropylene or polycarbonate syringe material.
Visually inspect Tecvayli for particulate matter and discolouration prior to administration. Do not use if the solution is discoloured, or cloudy, or if foreign particles are present.
Tecvayli solution for injection is colourless to light yellow.
Replace the transfer needle with an appropriately sized needle for injection.
Administration of Tecvayli.
Inject the required volume of Tecvayli into the subcutaneous tissue of the abdomen (preferred injection site). Alternatively, Tecvayli may be injected into the subcutaneous tissue at other sites (e.g. thigh). If multiple injections are required, Tecvayli injections should be at least 2 cm apart.
Do not inject into tattoos or scars or areas where the skin is red, bruised, tender, hard or not intact.
Any unused medicinal product or waste material should be disposed in accordance with local requirements.
Product is for single use in one patient only. Discard any residue.
Storage.
If Tecvayli is not used immediately, store at 2-8°C or up to 30°C for a maximum of 20 hours. Discard after 20 hours, if not used.
Monitoring.
Instruct patients to remain within proximity of a healthcare facility and monitor patients daily for 48 hours for signs and symptoms of CRS after administration of all doses within the Tecvayli step-up dosing schedule (see Table 1; Management of severe adverse reactions; see Section 4.4 Special Warnings and Precautions for Use, Cytokine release syndrome (CRS)).4.3 Contraindications
Hypersensitivity to the active substance(s) or to any of the excipients listed in Section 6.1.
4.4 Special Warnings and Precautions for Use
Cytokine release syndrome (CRS).
Cytokine release syndrome (CRS), including life-threatening or fatal reactions, may occur in patients receiving Tecvayli. In the MajesTEC-1 study, the median time to onset of CRS was 2 (Range: 1 to 6) days after the most recent dose with a median duration of 2 (Range: 1 to 9) days.
Clinical signs and symptoms of CRS may include, but are not limited to, fever, chills, hypotension, tachycardia, hypoxia, headache, and elevated liver enzymes. Potentially life-threatening complications of CRS may include cardiac dysfunction, adult respiratory distress syndrome, neurologic toxicity, renal and/or hepatic failure, and disseminated intravascular coagulation (DIC).
Initiate therapy according to Tecvayli step-up dosing schedule to reduce risk of CRS (see Table 1). Failure to follow the recommended doses or dosing schedule for initiation of therapy or re-initiation of therapy after dose delays may result in increased frequency and severity of adverse events related to mechanism of action.
Administer pretreatment medications (corticosteroids, antihistamine, and antipyretics) prior to each dose of the Tecvayli step-up dosing schedule to reduce risk of CRS and monitor patients following administration accordingly (see Section 4.2 Dose and Method of Administration, Pretreatment medications and Administration). In patients who experienced CRS following their previous dose, administer pretreatment medications prior to the next dose of Tecvayli.
Counsel patients to seek medical attention should signs or symptoms of CRS occur. A patient card to inform patients of CRS associated with Tecvayli is available. The patient card should be kept with the patient at all times whilst on treatment.
At the first sign of CRS, immediately evaluate patient for hospitalisation and institute treatment with supportive care, tocilizumab and/or corticosteroids, based on severity as indicated in Table 4.
In MajesTEC-1, tocilizumab, corticosteroids, and tocilizumab in combination with corticosteroids were used to treat 32%, 11% and 3% of CRS events, respectively. The use of myeloid growth factors, particularly granulocyte macrophage-colony stimulating factor (GM-CSF), should be avoided during CRS.
Withhold treatment with Tecvayli until CRS resolves as indicated in Table 3 (see Section 4.2 Dose and Method of Administration, Management of severe adverse reactions).
Neurologic toxicities.
Serious, life-threatening or fatal neurologic toxicities, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), occurred following treatment with Tecvayli. In MajesTEC-1, the majority of neurologic toxicity events were Grade 1 and Grade 2 (see Section 4.8 Adverse Effects (Undesirable Effects)). The onset of ICANS can be concurrent with CRS, following resolution of CRS, or in the absence of CRS. With longer follow-up, one event of seizure occurred in the context of an infectious toxicity (meningitis) in a patient who received Tecvayli.
Monitor patients for signs or symptoms of neurologic toxicities during treatment and treat promptly.
Counsel patients to seek medical attention should signs or symptoms of neurologic toxicity occur. At the first sign of neurologic toxicity, including ICANS, immediately evaluate patient and institute treatment based on severity as indicated in Table 5 (see Section 4.2 Dose and Method of Administration, Management of severe adverse reactions).
For ICANS or other neurologic toxicities, withhold treatment with Tecvayli as indicated in Table 3 and manage adverse reactions based on recommendations in Table 5.
Infections.
Severe, life-threatening or fatal infections have been reported in patients receiving Tecvayli (see Section 4.8 Adverse Effects (Undesirable Effects)). New or reactivated viral infections occurred during therapy with Tecvayli.
Monitor patients for signs and symptoms of infection prior to and during treatment with Tecvayli and treat appropriately. Prophylactic antimicrobials should be administered according to local institutional guidelines.
Withhold treatment with Tecvayli as indicated in Table 3 (see Section 4.2 Dose and Method of Administration, Dosage modifications).
Progressive multifocal leukoencephalopathy (PML), which can be fatal, has also been reported in patients receiving Tecvayli (see Section 4.8 Adverse Effects (Undesirable Effects)). Monitor any new onset of or changes in pre-existing neurological signs or symptoms. If PML is suspected, withhold treatment with Tecvayli and initiate appropriate diagnostic testing. Discontinue Tecvayli if PML is confirmed.
Hepatitis B virus reactivation.
Hepatitis B virus reactivation can occur in patients treated with drugs directed against B cells, and in some cases, may result in fulminant hepatitis, hepatic failure, and death.
Patients with evidence of positive HBV serology should be monitored for clinical and laboratory signs of HBV reactivation while receiving Tecvayli, and for at least six months following the end of treatment.
In patients who develop reactivation of HBV while on Tecvayli, withhold treatment with Tecvayli as indicated in Table 3 and manage per local institutional guidelines (see Section 4.2 Dose and Method of Administration, Dosage modifications).
Hypogammaglobulinaemia.
Hypogammaglobulinaemia has been reported in patients receiving Tecvayli (see Section 4.8 Adverse Effects (Undesirable Effects)).
Monitor immunoglobulin levels during treatment with Tecvayli and treat according to local institutional guidelines, including infection precautions, antibiotic or antiviral prophylaxis, and administration of immunoglobulin replacement therapy.
Vaccines.
Immune response to vaccines may be reduced when taking Tecvayli.
The safety of immunisation with live viral vaccines during or following Tecvayli treatment has not been studied. Vaccination with live virus vaccines is not recommended for at least 4 weeks prior to the start of treatment, during treatment, and at least 4 weeks after treatment.
Neutropenia.
Neutropenia and febrile neutropenia have been reported in patients who received Tecvayli (see Section 4.8 Adverse Effects (Undesirable Effects)).
Monitor complete blood cell counts at baseline and periodically during treatment and provide supportive care per local institutional guidelines.
Patients with neutropenia should be monitored for signs of infection.
Withhold treatment with Tecvayli based on severity as indicated in Table 3 (see Section 4.2 Dose and Method of Administration, Dosage modifications).
Use in the elderly (65 years of age and older).
Of the 165 patients treated with Tecvayli in MajesTEC-1 at the recommended dose, 48% were 65 years of age or older, and 15% were 75 years of age or older. No overall differences in safety or effectiveness were observed between these patients and younger patients. No dose adjustment is necessary (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Paediatric use (17 years of age and younger).
The safety and efficacy of Tecvayli have not been established in paediatric patients aged 17 years and younger.
Effects of laboratory tests.
No data available.
Effect on QT/QTc interval and cardiac electrophysiology.
At the recommended treatment dose (1.5 mg/kg) of Tecvayli, no clinically relevant QTc prolongation has been observed.4.5 Interactions with Other Medicines and Other Forms of Interactions
No drug interaction studies have been performed with Tecvayli.
The initial release of cytokines associated with the start of Tecvayli treatment could suppress CYP450 enzymes. Based on physiologically based pharmacokinetic (PBPK) modelling, the highest risk of drug-drug interaction is predicted to be from initiation of Tecvayli step-up dosing schedule up to 7 days after the first treatment dose or during a CRS event. During this time period, monitor for toxicity or drug concentrations (e.g. cyclosporine) in patients who are receiving concomitant CYP450 substrates with a narrow therapeutic index. The dose of the concomitant drug should be adjusted as needed.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the effect of Tecvayli on fertility.
No studies have been conducted to evaluate the effects of teclistamab on fertility in males or females.
(Category C)
There are no available data on the use of Tecvayli in pregnant women or animal data to assess the risk of Tecvayli in pregnancy. Teclistamab causes T-cell activation and cytokine release; immune activation may compromise pregnancy maintenance. Human IgG is known to cross the placenta after the first trimester of pregnancy.
Therefore, teclistamab has the potential to be transmitted from the mother to the developing fetus.
Tecvayli is not recommended for women who are pregnant. Tecvayli is associated with hypogammaglobulinaemia, therefore, assessment of immunoglobulin levels in newborns of mothers treated with Tecvayli should be considered.
Pregnancy testing.
Pregnancy status for females of child-bearing potential should be verified prior to starting treatment with Tecvayli.
Contraception.
Advise females of reproductive potential to use effective contraception during treatment and for five months after the final dose of Tecvayli.
Advise male patients with a female partner of reproductive potential to use effective contraception during treatment and for three months after the last dose of Tecvayli.
It is not known whether teclistamab is excreted in human or animal milk, affects breastfed infants or affects milk production. Because of the potential for serious adverse reactions in breastfed infants from Tecvayli, advise patients not to breastfeed during treatment with Tecvayli and for at least five months after the last dose.4.7 Effects on Ability to Drive and Use Machines
Due to the potential for ICANS, patients receiving Tecvayli are at risk of depressed level of consciousness. Patients should avoid driving or operating heavy or potentially dangerous machinery during and for 48 hours after completion of Tecvayli step-up dosing schedule and in the event of new onset of any neurological symptoms (Table 1) (see Section 4.2 Dose and Method of Administration).
4.8 Adverse Effects (Undesirable Effects)
The safety data of Tecvayli was evaluated in MajesTEC-1, which included 165 adult patients with relapsed or refractory multiple myeloma who received the recommended dose regimen of subcutaneous Tecvayli as monotherapy. The median duration of Tecvayli treatment was 8.5 (range: 0.2 to 24.4) months.
The most frequent adverse reactions of any grade (≥ 20%) in patients were hypogammaglobulinaemia (75%), cytokine release syndrome (72%), neutropenia (71%), anaemia (55%), musculoskeletal pain (52%), fatigue (41%), thrombocytopenia (40%), injection site reaction (38%), upper respiratory tract infection (37%), lymphopenia (35%), diarrhoea (28%), pneumonia (28%), nausea (27%), pyrexia (27%), headache (24%) cough (24%), constipation (21%), and pain (21%).
Serious adverse reactions were reported in 65% patients who received Tecvayli. Serious adverse reactions reported in ≥ 2% of patients included pneumonia (16%), COVID-19 (15%), cytokine release syndrome (8%), sepsis (7%), pyrexia (5%), musculoskeletal pain (5%), acute kidney injury (4.8%), diarrhoea (3.0%), cellulitis (2.4%), hypoxia (2.4%), febrile neutropenia (2.4%), and encephalopathy (2.4%).
Dose interruptions (dose delays and dose skips) of Tecvayli due to adverse reactions occurred in 65% of patients. The most frequent adverse reactions (≥ 5%) leading to dose interruptions were neutropenia (26%), COVID-19 (12%), pneumonia (10%), cytokine release syndrome (8%) and pyrexia (7%).
Dose reduction of Tecvayli due to adverse reaction occurred in one patient (0.6%) due to neutropenia.
Permanent discontinuation of Tecvayli due to adverse reactions occurred in two patients (1.2%), both due to infection.
Table 9 summarises adverse reactions reported in patients who received Tecvayli.
Adverse reactions observed during clinical studies are listed below by frequency category. Frequency categories are defined as follows: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1000 to < 1/100); rare (≥ 1/10,000 to < 1/1000); very rare (< 1/10,000); not known (frequency cannot be estimated from the available data).
Within each frequency grouping, undesirable effects are presented in order of decreasing frequency.

Description of selected adverse reactions.
Cytokine release syndrome.
In Majes-TEC-1 (N=165), CRS was reported in 72% of patients following treatment with Tecvayli. One-third (33%) of patients experienced more than one CRS event. Most patients experienced CRS following step-up dose 1 (44%), step-up dose 2 (35%), or the initial treatment dose (24%). Less than 3% of patients developed first occurrence of CRS following subsequent doses of Tecvayli. Most CRS events were Grade 1 (50%) and Grade 2 (21%). Less than one percent (0.6%) of CRS events were Grade 3, and no Grade 4 or fatal events occurred. Tocilizumab, corticosteroids, and tocilizumab in combination with corticosteroids were used to treat 32%, 11% and 3% of CRS events, respectively.
The most frequent (≥ 3%) signs and symptoms associated with CRS were fever (72%), hypoxia (13%), chills (12%), hypotension (12%), sinus tachycardia (7%), headache (7%), and elevated liver enzymes (aspartate aminotransferase and alanine aminotransferase elevation) (3.6% each).
Neurologic toxicities.
In Majes-TEC-1 (N=165), neurologic toxicities were reported in 15% of patients receiving Tecvayli. Most neurologic toxicity events were Grade 1 (8.5%), Grade 2 (5.5%) and Grade 4 (0.6%). The most frequently reported neurologic toxicity was headache (8.5%).
ICANS was reported in 3% of patients receiving Tecvayli at the recommended dose. The most frequent clinical manifestations of ICANS reported were confusional state (1.2%) and dysgraphia (1.2%).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Symptoms and signs.
The maximum tolerated dose of teclistamab has not been determined. In clinical trials, doses of up to 6 mg/kg have been administered.
Treatment.
In the event of an overdose, the patient should be monitored for any signs or symptoms of adverse effects and appropriate symptomatic treatment should be instituted immediately.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: other monoclonal antibodies and antibody drug conjugates, ATC code: L01FX24.
Within the first month of treatment with teclistamab, activation and redistribution of T-cells, reduction of B-cells and induction of serum cytokines were observed.
Within one month, the majority of responders had reduction in soluble BCMA, and a greater reduction in soluble BCMA was observed in patients with deeper responses to teclistamab.
Immunogenicity.
Patients treated with subcutaneous teclistamab monotherapy (N=238) in MajesTEC-1 were evaluated for antibodies to teclistamab using an electrochemiluminescence-based immunoassay. One patient (0.4%) developed antibodies to teclistamab of low titre which were neutralising.
Mechanism of action.
Teclistamab is a bispecific antibody that targets the CD3 receptor expressed on the surface of T cells and B cell maturation antigen (BCMA), which is expressed on the surface of malignant multiple myeloma B-lineage cells, as well as late-stage B cells and plasma cells.
In vitro, teclistamab activated T-cells, caused the release of various proinflammatory cytokines, and resulted in the lysis of multiple myeloma cells.
Clinical trials.
The efficacy of Tecvayli monotherapy was evaluated in patients with relapsed or refractory multiple myeloma in a single-arm, open-label, multicentre, study (MajesTEC-1 (MMY1001)). The study included patients who had previously received at least three prior therapies, including a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody. The study excluded patients who experienced stroke or seizure within the past 6 months and patients with active or documented history of autoimmune disease, with the exception of vitiligo, type 1 diabetes and prior autoimmune thyroiditis.
Patients received initial step-up doses of 0.06 mg/kg and 0.3 mg/kg of Tecvayli administered subcutaneously followed by the treatment dose of Tecvayli 1.5 mg/kg administered subcutaneously once weekly thereafter until disease progression or unacceptable toxicity (see Section 4.2 Dose and Method of Administration, Dosage - adults (18 years of age and older)).
Patients who had a complete response (CR) or better for a minimum of 6 months were eligible to reduce dosing frequency to 1.5 mg/kg subcutaneously every two weeks until disease progression or unacceptable toxicity (see Section 4.2 Dose and Method of Administration, Dosage - adults (18 years of age and older)).
The median duration between step-up dose 1 and step-up dose 2 was 2.9 (range: 2-7) days. The median duration between step-up dose 2 and the initial treatment dose was 3.9 (range: 2-9) days. Patients were hospitalised for monitoring for at least 48 hours after administration of each dose of the Tecvayli step-up dosing schedule.
The efficacy population included 165 patients. The median age was 64 (range: 33-84) years with 15% of patients ≥ 75 years of age; 58% were male; 81% were White, 13% were Black, 2% were Asian. The International Staging System (ISS) at study entry was 52% in Stage I, 35% in Stage II, and 12% in Stage III. High-risk cytogenetics (presence of del(17p), t(4;14) or t(14; 16)) were present in 26% of patients. Seventeen percent (17%) of patients had extramedullary plasmacytomas.
The median time since initial diagnosis of multiple myeloma to enrolment was 6 (range: 0.8 22.7) years. The median number of prior therapies was 5 (range: 2-14) with 23% of patients who received 3 prior lines of therapy. Eighty-two percent of patients received prior stem cell transplantation. All patients had received prior therapy with a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody, and 78% were triple-class refractory (refractory to PI, an IMiD agent and an anti-CD38 monoclonal antibody).
Efficacy results were based on overall response rate as determined by the Independent Review Committee (IRC) assessment using International Myeloma Working Group (IMWG) 2016 criteria (see Table 10 and Table 11).


5.2 Pharmacokinetic Properties
The Cmax and AUCtau of teclistamab after the first subcutaneous treatment dose increase proportionally over a dosage range of 0.08 mg/kg to 3 mg/kg (0.05 to 2 times the approved recommended treatment dosage). Ninety percent of steady state exposure was achieved after 12 weekly treatment doses. The mean accumulation ratio between the first and 13th weekly treatment dose of teclistamab 1.5 mg/kg was 4.2-fold for Cmax, 4.1-fold for Ctrough, and 5.3-fold for AUCtau. See Table 12.

Absorption.
The mean bioavailability of teclistamab was 72% when administered subcutaneously. The median (range) Tmax of teclistamab after the first and 13th weekly treatment doses were 139 (19 to 168) hours and 72 (24 to 168) hours, respectively.
Distribution.
Based on the population pharmacokinetic model, mean volume of distribution was 5.63 L (29% coefficient of variation (CV)).
Metabolism and excretion.
Population pharmacokinetic analysis showed that teclistamab clearance decreases over time, with a mean (CV%) maximal reduction from baseline to the 13th weekly treatment dose of 40.8% (56%). The geometric mean (CV%) clearance is 0.472 L/day (64%) at the 13th weekly treatment dose. Patients who discontinue teclistamab after the 13th weekly treatment dose are expected to have a 50% reduction from Cmax in teclistamab concentration at a median (5th to 95th percentile) time of 15 (7 to 33) days after Tmax and a 97% reduction from Cmax in teclistamab concentration at a median time of 69 (32 to 163) days after Tmax.
Population pharmacokinetic analysis (based on MajesTEC-1) showed that soluble BCMA levels did not impact teclistamab serum concentrations.
Special populations.
Renal impairment.
No formal studies of Tecvayli in patients with renal impairment have been conducted.
Results of population pharmacokinetic analyses indicate that mild (60 mL/min/1.73 m2 ≤ estimated glomerular filtration rate (eGFR) < 90 mL/min/1.73 m2) or moderate (30 mL/min/1.73 m2 ≤ eGFR < 60 mL/min/1.73 m2) renal impairment did not significantly influence the pharmacokinetics of teclistamab. Limited data are available from patients with severe renal impairment.
Hepatic impairment.
No formal studies of Tecvayli in patients with hepatic impairment have been conducted.
Results of population pharmacokinetic analyses indicate that mild hepatic impairment (total bilirubin > 1 to 1.5 times upper limit of normal (ULN) and any aspartate aminotransferase (AST), or total bilirubin ≤ ULN and AST > ULN) did not significantly influence the pharmacokinetics of teclistamab. No data are available in patients with moderate and severe hepatic impairment.
Age and sex.
The pharmacokinetics of Tecvayli in paediatric patients have not been investigated.
Results of population pharmacokinetic analyses indicate that age (24 to 84 years) and sex did not influence the pharmacokinetics of teclistamab.
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been performed to assess the genotoxic potential of teclistamab. As a large protein molecule, teclistamab is not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity.
No carcinogenicity studies have been performed to assess the carcinogenic potential of teclistamab.6 Pharmaceutical Particulars
6.1 List of Excipients
Disodium edetate (EDTA), glacial acetic acid, polysorbate 20, sodium acetate trihydrate, sucrose, water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store at 2°C to 8°C. Refrigerate, do not freeze.
Store in the original carton in order to protect from light.
6.5 Nature and Contents of Container
3 mL solution for injection in a Type 1 glass vial with an elastomeric closure and aluminium seal with a flip off button containing 30 mg of sterile teclistamab (10 mg/mL). Pack size of 1 vial.
1.7 mL solution for injection in a Type 1 glass vial with an elastomeric closure and aluminium seal with a flip off button containing 153 mg of sterile teclistamab (90 mg/mL). Pack size of 1 vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
CAS number.
2119595-80-9.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes
