What is in this leaflet
Read this leaflet carefully before you start using this medicine. This leaflet answers some common questions about TEGLUTIK® oral suspension.
It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you using TEGLUTIK® oral suspension against the benefits they expect it will have for you.
If you have any concerns about using this medicine, talk to your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What TEGLUTIK® is used for
The brand name of your medicine is TEGLUTIK®. The active ingredient in the medicine is called riluzole, which acts on the nervous system.
TEGLUTIK® is used to treat people with amyotrophic lateral sclerosis (ALS), a form of motor neurone disease, which can cause muscle degeneration leading to muscle weakness.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
TEGLUTIK® oral suspension is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
It is important to remember that you may not feel any different when you take TEGLUTIK®. The benefits of using TEGLUTIK® may not be noticeable to you. You should not stop taking TEGLUTIK® without speaking to your doctor first.
Before you take it
When you must not use it
Do not use TEGLUTIK® if you:
- have liver disease
- are pregnant or intend to become pregnant
- are breastfeeding or intend to breastfeed
Do not use this medicine if you are allergic to riluzole or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not give this medicine to a child. There is no experience with the use of this medicine in children.
Do not take this medicine if the expiry date (EXP) printed on the pack has passed. If you take it after the expiry date has passed, it may not work as well.
Do not take this medicine if the packaging is torn or shows signs of tampering.
Before you start to use it
Tell your doctor if you are allergic to any of the ingredients listed at the end of this leaflet.
Tell your doctor if you are pregnant or intend to become pregnant. This medicine is not recommended to be used during pregnancy.
Tell your doctor if you are breastfeeding or planning to breastfeed. It is not known whether it passes into breast milk. Your doctor will discuss the risks and benefits of taking it if you are breastfeeding or planning to breastfeed.
Tell your doctor if you have or have had any medical conditions, especially the following:
- liver disease
- kidney disease
- lung disease
Tell your doctor if you plan to have surgery.
If you have not told your doctor about any of the above, do so before you take this medicine.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines and TEGLUTIK® may interfere with each other. These include:
- theophylline - a medicine used to treat asthma
- amitriptyline - a medicine used to treat depression
- tacrine - a medicine used in patients with Alzheimer's Disease
- some types of antibiotics eg. rifampicin and quinolones
- omeprazole - a medicine used to treat gastric ulcers.
- some medicines used to treat depression eg. Clomipramine and fluvoxamine
- diazepam - a medicine for sedation.
- diclofenac - a medicine used to reduce pain and inflammation.
These medicines may be affected by TEGLUTIK®, or may affect how well it works. You may need to use different amounts of your medicine or you may need to take different medicines. Your doctor or pharmacist will advise you.
Tell your doctor if you smoke and how much coffee you drink. Nicotine and caffeine may affect the amount of TEGLUTIK® in your body.
TEGLUTIK® contains liquid sorbitol (E420). If you have been told by your doctor that you have an intolerance to some sugars, contact your doctor before taking this medicinal product.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take TEGLUTIK®
Follow all directions given to you by your doctor or pharmacist carefully.
If you do not understand the instructions in this leaflet, ask your doctor or pharmacist for help.
How much to use
The recommended dose is 100 mg a day (50 mg every 12 hours).
Do not take more than the dose your doctor has directed.
How to take TEGLUTIK®
10 mL of the oral suspension, containing 50 mg of riluzole, should be taken by mouth or via a Percutaneous Endoscopic Gastrostomy (PEG) every 12 hours, at the same time of the day each day (for example, in the morning and evening). The suspension is administered by means of graduated dosing syringe.
The oral suspension must be manually gently shaken for at least 30 seconds by rotating the bottle by 180° and the homogeneity should be visually verified.
Method of administration:
Open the bottle: press the cap and turn it anticlockwise (figure 1).
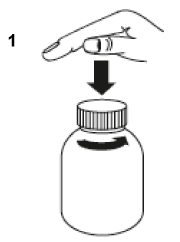
Take the syringe, remove the tip and insert the syringe in the adaptor opening (figure 2). Turn the bottle upside down (figure 3).
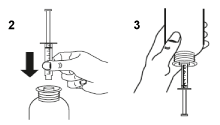
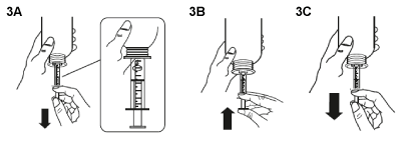
Fill the syringe with a small amount of suspension by pulling the plunger down (figure 3A), then push the piston upward in order to remove any possible bubble (figure 3B). Pull the piston down to the graduation mark corresponding to the quantity in milliliters (mL) prescribed by your doctor (figure 3C).
Turn the bottle the right way up (figure 4A). Remove the syringe from the adaptor (figure 4B).
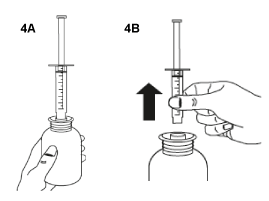
- Administer orally the whole content of the syringe. Dilution in water is not necessary.
- Close the bottle with the plastic screw cap.
- Wash the syringe with water only and re-assemble it with its tip cap once dried (figure 5).
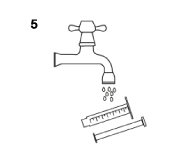
For PEG administration, the tubing should be washed with 10 mL of water before and after administration of TEGLUTIK®.
When to take it
10 mL of the oral suspension should be taken by mouth every 12 hours, at the same time of the day each day (for example, in the morning and evening).
TEGLUTIK® should not be taken immediately before or after meals, especially meals which may contain food high in fat. TEGLUTIK® may not work as well if it is taken at the same time as your meals.
How long to take it
Do not stop taking TEGLUTIK® unless your doctor tells you to, even if you feel better.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take the next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not use a double dose to make up for the dose you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you use too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre (telephone 13 11 26), or go to the Accident and Emergency at your nearest hospital, if you think you or anyone else may have used too much TEGLUTIK® oral suspension. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking TEGLUTIK®
Things you must do
Tell all the doctors, dentists and pharmacists who are treating you that you are taking TEGLUTIK®.
If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking TEGLUTIK®.
If you plan to have surgery that needs a general anaesthetic, tell your doctor or dentist that you are taking this medicine. If you become pregnant while you are taking this medicine tell your doctor immediately.
During your treatment with TEGLUTIK® your doctor will do some blood tests from time to time to check for any possible signs of liver damage.
Things you must not do
Do not take more than the recommended dose unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not use this medicine to treat any other complaints unless your doctor tells you to.
Do not stop taking this medicine, or lower the dosage, without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how this medicine affects you. This medicine may cause drowsiness or dizziness in some people. Make sure you know how you react to it before you drive a car, operate machinery, or do anything else that could be dangerous if you are dizzy.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TEGLUTIK®.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following:
- stomach ache, nausea or vomiting
- headache
- joint stiffness
- skin problems eg. rash, flaking skin
- dizziness
- sleepiness
- weakness or loss of strength
These are the most common side effects of this medicine.
Tell your doctor immediately, or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- irregular or fast heartbeat
- frequent infections such as fever, severe chills, sore throat or mouth ulcers
- swelling of the hands, feet or legs
- tingling sensations around the mouth
- shortness of breath or difficulty breathing
These may be serious side effects of TEGLUTIK®. You may need urgent medical attention. Serious side effects are rare.
If any of the following happen, stop taking this medicine and tell your doctor immediately, or go to Accident and Emergency at your nearest hospital:
- Severe upper stomach pain, often with nausea and vomiting
- If your skin becomes itchy or yellow or if you start to bleed or bruise easily. You may be developing a liver problem.
These are very serious side effects. You may need urgent medical attention or hospitalisation. All of these side effects are very rare.
As riluzole oral suspension is more rapidly absorbed than riluzole tablets, a slight increase in tiredness, dizziness, diarrhoea and transaminases cannot be excluded. If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet.
Other side effects not listed above may also occur in some patients. Tell your doctor if you notice anything that is making you feel unwell.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using TEGLUTIK®
Storage
Keep the oral suspension in the bottle, inside the box, until it is time to take it. If you take the oral suspension out of the box or bottle it may not keep well
Keep it in a cool dry place where the temperature stays below 25°C.
Do not store it or any other medicine in the bathroom or near a sink.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
After first opening: the oral suspension should be used with 15 days.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking TEGLUTIK®, or it has passed its expiry date, ask your pharmacist what to do with any that are left over.
Product description
What it looks like
This medicine is presented as a slightly brown, opaque homogeneous oral suspension after being manually gently shaken.
TEGLUTIK® is available in a carton containing a bottle of 250 mL or 300 mL, or in a multipack consisting of 2 single cartons each with one bottle of 250 mL. Each carton is supplied with a plastic graduated oral dosing syringe. The syringe barrel is graduated in millilitres up to 10 mL. Not all pack sizes may be marketed.
Ingredients
The active substance is riluzole. 1 mL of oral suspension contains 5 mg of riluzole.
The oral suspension also contains:
- sorbitol solution (70% w/w) (non-crystallising)
- aluminium magnesium silicate
- xanthan gum
- saccharin sodium
- antifoam AF emulsion Q7-2587
- sodium lauryl sulfate
- ceteareth-25
- purified water
1 mL of oral suspension contains 400 mg of sorbitol (equivalent to 571.43 mg of sorbitol solution (70% w/w)). 20 mL daily dose of suspension contains 8 g of sorbitol.
Products containing sorbitol may have a laxative effect or cause diarrhoea.
TEGLUTIK® is distributed by:
Seqirus Pty Ltd
ABN: 26 160 735 035
63 Poplar Road
Parkville
Victoria 3052
Telephone: 1800 642 865
www.seqirus.com.au
Australian registration numbers: AUST R292987
Date of preparation: April 2021
TEGLUTIK® is a registered trade mark of Italfarmaco S.A., Spain
Published by MIMS November 2021
 The adverse events reported in the comparative bioavailability study between Teglutik oral suspension and riluzole tablets are reported by body system in Table 2:
The adverse events reported in the comparative bioavailability study between Teglutik oral suspension and riluzole tablets are reported by body system in Table 2: A slightly higher risk of the adverse events considered related to Cmax (i.e. ~20% higher than riluzole tablets) of Teglutik oral suspension (e.g. dizziness, diarrhoea, asthenia and ALT increase) cannot be excluded.
A slightly higher risk of the adverse events considered related to Cmax (i.e. ~20% higher than riluzole tablets) of Teglutik oral suspension (e.g. dizziness, diarrhoea, asthenia and ALT increase) cannot be excluded. In a second dose-ranging trial, 959 patients with ALS were randomised to one of four treatment groups: riluzole 50, 100, 200 mg/day, or placebo and were followed-up for 18 months. In patients treated with riluzole 100 mg/day, survival was significantly longer compared to patients who received placebo. The median survival time approached 16.5 months versus 13.5 months for riluzole 100 mg/day and placebo, respectively. There were no changes from baseline observed in the functional evaluation. The effect of 50 mg/day was not statistically significant compared to placebo and the effect of 200 mg/day was essentially comparable to that of 100 mg/day. See Figure 2.
In a second dose-ranging trial, 959 patients with ALS were randomised to one of four treatment groups: riluzole 50, 100, 200 mg/day, or placebo and were followed-up for 18 months. In patients treated with riluzole 100 mg/day, survival was significantly longer compared to patients who received placebo. The median survival time approached 16.5 months versus 13.5 months for riluzole 100 mg/day and placebo, respectively. There were no changes from baseline observed in the functional evaluation. The effect of 50 mg/day was not statistically significant compared to placebo and the effect of 200 mg/day was essentially comparable to that of 100 mg/day. See Figure 2. A separate compassionate-use study (n=168), enabling access to treatment for patients excluded from the two pivotal studies, was designed to assess the efficacy and safety of riluzole in patients at a late stage of the disease. In this population with decreased respiratory function (baseline vital capacity less than 60%), survival time and motor function in the riluzole group did not differ significantly from that of placebo. It was anticipated that up to 300 patients would enter this study, but only 168 were enrolled (86 received placebo, 82 received riluzole). Thus the statistical power of the study was diminished.
A separate compassionate-use study (n=168), enabling access to treatment for patients excluded from the two pivotal studies, was designed to assess the efficacy and safety of riluzole in patients at a late stage of the disease. In this population with decreased respiratory function (baseline vital capacity less than 60%), survival time and motor function in the riluzole group did not differ significantly from that of placebo. It was anticipated that up to 300 patients would enter this study, but only 168 were enrolled (86 received placebo, 82 received riluzole). Thus the statistical power of the study was diminished.
