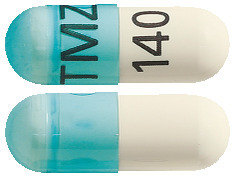


What is in this leaflet
This leaflet answers some common questions about TEMIZOLE. It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking TEMIZOLE against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, talk to your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What TEMIZOLE is used for
This medicine contains temozolomide as the active ingredient.
TEMIZOLE is used to treat:
- patients with brain tumours
- adult patients with advanced metastatic malignant melanoma
It belongs to a group of medicines called cytotoxic or chemotherapy medicines.
TEMIZOLE works by killing cancer cells and stopping cancer cells from growing and multiplying.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
Use in children
TEMIZOLE can be used to treat children aged 3 years and older, with specific forms of brain cancer (glioblastoma multiforme or anaplastic astrocytoma, showing reccurrence or progression after standard therapy).
Before you take TEMIZOLE
When you must not take it
Do not take TEMIZOLE if you have an allergy to:
- any medicine containing temozolomide
- any medicine containing dacarbazine
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take TEMIZOLE if you or your partner are pregnant or intend to become pregnant. TEMIZOLE may cause birth defects if either the male or female is taking this medicine at the time of conception or during pregnancy.
Female patients must have a negative pregnancy test before starting TEMIZOLE.
Both male and female patients and their partners should each use birth control whilst taking TEMIZOLE.
Male patients whose partners are already pregnant should use a condom to minimise exposure to the unborn baby by TEMIZOLE in the sperm.
Do not breast-feed if you are taking this medicine.
Do not take TEMIZOLE if you have very low levels of white blood cells, red blood cells or platelets.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- liver or kidney problems
TEMIZOLE could cause hepatitis B to become active again, which can be fatal in some cases. - anaemia or blood clotting problems
Tell your doctor if you intend to have children. TEMIZOLE may cause infertility in men.
Tell your doctor if you vomit frequently. Your doctor may give you other medication to control the vomiting.
If you have not told your doctor about any of the above, tell them before you start taking TEMIZOLE.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and TEMIZOLE may interfere with each other. These include other medicines used to treat cancer or any other treatments that may affect your immune system.
You may need different amounts of your medicines or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take TEMIZOLE
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the bottle, ask your doctor or pharmacist for help.
How much to take
Your doctor has worked out the exact dose of TEMIZOLE for you according to your individual needs.
You may be given other medication to take before or after TEMIZOLE to help stop nausea.
TEMIZOLE in combination treatment with radiation (newly diagnosed patients)
If you are a patient with a newly diagnosed brain tumour, your doctor will start you on a dose of TEMIZOLE every day for 42 days (up to 49 days) in combination with radiation therapy. This is the first part of the treatment ("concomitant phase") in which you complete the radiation therapy. Your treatment will be interrupted for 4 weeks to give your body a chance to recover.
You will then start the next phase of treatment ("adjuvant phase") and your TEMIZOLE dose will change. In this phase, there are up to 6 treatment cycles. Each treatment cycle lasts 28 days. You will take your new dose of TEMIZOLE once a day for the first five days ("dosing days") of each cycle, followed by 23 days without TEMIZOLE. This makes a 28 day treatment cycle.
After day 28, the next cycle will begin, in which you will again take this medicine once daily for five days followed by 23 days without TEMIZOLE.
Before each new treatment cycle begins, your blood will be tested to determine if the TEMIZOLE dose needs to be adjusted.
TEMIZOLE alone (patients treated for recurrent brain tumour)
Take the dose your doctor has prescribed once a day for five days.
Depending on your response to TEMIZOLE, a new treatment cycle will begin each 28 days. You will then take this medicine again once daily for five days.
Before each new treatment cycle, your blood will be tested to determine if the TEMIZOLE dose needs to be changed.
How to take it
Before you start each new treatment cycle, be sure you understand exactly how many capsules of each strength you need to take on each day of dosing.
Swallow the capsules whole with a glass of water.
Do not open or chew the capsules.
TEMIZOLE comes in different strengths (shown on the label in ‘mg’). Each strength is a different colour. Depending on your dose of TEMIZOLE, you may have to take several capsules on each dosing day of the treatment cycle
Be sure you understand exactly how many capsules you need to take of each strength.
Ask your doctor or pharmacist to write down the number of each strength (include colour) that you need to take on each dosing day.
Be sure you know exactly which days are your dosing days.
Be sure you review the dose with your doctor each time you start a new treatment cycle. Sometimes the dose or the mix of capsules you need to take will be different from the last cycle.
Once you take this medicine home, if you are confused or unsure about how to take your dose, call your doctor or pharmacist before beginning the treatment cycle. Errors in how you take this medicine may have serious health consequences.
When to take it
Take your medicine on an empty stomach at least one hour before a meal.
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
If vomiting occurs after you take your medicine, do not take another dose that day.
How long to take it
Continue taking your medicine for as long as your doctor tells you. Your doctor will tell you when your treatment should be stopped.
If you forget to take it
If you miss a dose, take the missed dose as soon as possible during the same day. If a full day has gone by, check with your doctor.
Do not take a double dose to make up for the dose you missed unless your doctor tells you to.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think you or anyone else may have taken too much TEMIZOLE. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using TEMIZOLE
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking TEMIZOLE.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
Tell your doctor if you feel sick or vomit while being treated with TEMIZOLE. Your doctor may give you another medicine to help with this.
Tell your doctor if you become unusually pale or tired, get blood clotting problems or frequent infections while being treated with TEMIZOLE. This could be caused by a low level of red blood cells, platelets or white blood cells in your blood.
This is more common in patients over 70 years of age.
Your doctor may need to change your dose of TEMIZOLE.
Tell your doctor immediately if you or your partner becomes pregnant while taking TEMIZOLE.
Be sure to keep all your doctor's appointments so your progress can be checked. Your doctor may need to do some blood and other tests from time to time to check on your progress and detect any unwanted side effects.
Keep follow-up appointments with your doctor. It is important to have your follow-up doses of TEMIZOLE at the appropriate times to get the best effects from your treatment.
Things you must not do
Do not take TEMIZOLE to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not open the capsules. If a capsule is damaged, avoid contact with your skin, eyes and nose. Avoid inhaling the powder. If you touch the powder or get some in your eyes or nose, wash the area with water.
Do not stop taking your medicine or change the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how TEMIZOLE affects you. This medicine may cause drowsiness in some people. If this occurs, do not drive, operate machinery or do anything else that could be dangerous. Children should be careful when riding bicycles or climbing trees.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TEMIZOLE.
Like other medicines used in the treatment of cancer, TEMIZOLE may have unwanted side effects. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- nausea, vomiting, feeling unwell
- tiredness, sleepiness
- constipation
- headache
- loss of appetite or weight
- diarrhoea
- fever or high temperature
- rash, hair loss, itching
- dizziness, weakness
- general body pain
- stomach pain, indigestion
- different taste sensation
- mouth ulcers
- coughing
- sleeplessness
The above list includes the more common side effects of your medicine.
Tell your doctor as soon as possible if you notice any of the following:
- shortness of breath
- tingling or numbness in hands or feet
- bruising, bleeding or being unusually pale or tired
This could be caused by a low level of platelets or red blood cells in the blood. - new or recurring cytomegalovirus infection and return of hepatitis B
- symptoms of diabetes including passing a large amount of urine and constant thirst
- symptoms such as fever, headache, personality changes, seizures and/or vomiting which could be associated with a brain infection caused by the herpes virus
- shivering that is associated with chills and fever
This could be a sign of an infection caused by a low level of white blood cells in the blood. - development of red or purple spots under the skin
Shivering associated with chills and fever or the development of red or purple spots under the skin may take some time to occur.
Therefore, even after you have finished your treatment with TEMIZOLE, you should tell your doctor immediately if you notice these side effects.
The above list includes serious side effects that may require medical attention.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using TEMIZOLE
Storage
Keep your capsules in the bottle until it is time to take them. If you take the capsules out of the bottle they may not keep well.
Keep your capsules in a cool dry place where the temperature stays below 25C.
Do not store TEMIZOLE or any other medicine in the bathroom or near a sink. Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
TEMIZOLE comes in 5 strengths:
- TEMIZOLE 5 mg – Green/White hard gelatin capsules, size 3, imprinted 'TMZ' on the cap and '5' on the body containing white to light pink powder.
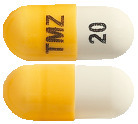
- TEMIZOLE 20 mg – Yellow/ White hard gelatin capsules, size 5, imprinted 'TMZ' on the cap and '20' on the body containing white to light pink powder.
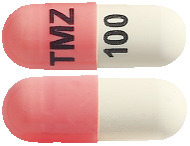
- TEMIZOLE 100 mg – Pink/ White hard gelatin capsules, size 3, imprinted 'TMZ' on the cap and '100' on the body containing white to light pink powder.
- TEMIZOLE 140 mg – Transparent blue/White hard gelatin capsules, size 1, imprinted 'TMZ' on the cap and '140' on the body containing white to light pink powder.
- TEMIZOLE 250 mg – White/ White hard gelatin capsules, size 0, imprinted 'TMZ' on the cap and '250' on the body containing white to light pink powder.
Each bottle contains 5 capsules.
Ingredients
TEMIZOLE contains either 5 mg, 20 mg, 100 mg, 140 mg or 250 mg of temozolomide as the active ingredient.
The capsules also contain the following inactive ingredients:
- colloidal anhydrous silica
- lactose
- sodium starch glycollate
- stearic acid
- tartaric acid
The capsule shells contain:
- gelatin
- indigo carmine (5 mg and 140 mg capsules only)
- iron oxide red (100 mg capsules only)
- iron oxide yellow (5 mg and 20 mg capsules only)
- purified water
- sodium lauryl sulfate (250 mg capsules only)
- titanium dioxide
The ink present on the capsules is TekPrint SW-9008 black ink (ARTG PI No: 2328).
TEMIZOLE contains sulfites and sugars as lactose.
Distributor
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in February 2022.
Australian registration numbers:
TEMIZOLE 5mg - AUST R 172973
TEMIZOLE 20mg - AUST R 172968
TEMIZOLE 100mg - AUST R 172964
TEMIZOLE 140mg - AUST R 172972
TEMIZOLE 250mg - AUST R 172975
TEMIZOLE® is a Viatris company trade mark
TEMIZOLE_cmi\Feb22/00
Published by MIMS April 2022





 Temozolomide is a white to pale brown/pink, crystalline powder which is odourless or almost odourless and hygroscopic. It is slightly soluble in water (3.1 mg/mL), methanol (4.4 mg/mL) and ethanol (0.6 mg/mL).
Temozolomide is a white to pale brown/pink, crystalline powder which is odourless or almost odourless and hygroscopic. It is slightly soluble in water (3.1 mg/mL), methanol (4.4 mg/mL) and ethanol (0.6 mg/mL).