What is in this leaflet
This leaflet answers some common questions about tenofovir. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may want to read it again.
What this medicine is used for
Tenofovir belongs to a group of medicines called nucleoside analog reverse transcriptase inhibitors (NRTIs).
Chronic Hepatitis B (CHB)
Tenofovir is an antiviral medication used to treat CHB in adults and children aged over 12 years and weighing at least 35 kg.
Tenofovir works by interfering with the normal working of enzymes that are essential for HBV to reproduce itself. This may help lower the amount of hepatitis B virus in your body by lowering the ability of the virus to multiply and infect new liver cells. It can improve the inflammation and scar tissue caused by the hepatitis B virus in your liver. Lowering the amount of virus in your body may reduce the chance of developing cirrhosis, liver failure and liver cancer.
We do not know how long this medicine may help treat your hepatitis. Sometimes viruses change in your body and medicines no longer work. This is called drug resistance.
Human Immunodeficiency Virus (HIV-1) Infection
Tenofovir is also used to treat HIV-1 infection in adults and children aged over 12 years and weighing at least 35 kg. It is always used in combination with other anti-HIV-1 medicines to treat people with HIV-1 infection.
HIV-1 infection destroys CD4 (T) cells, which are important to the immune system. After many T cells are destroyed, acquired immune deficiency syndrome (AIDS) develops.
This medicine helps to block HIV-1 reverse transcriptase, an enzyme in your body that is needed for HIV-1 to multiply. It lowers the amount of HIV-1 in the blood (called viral load) and may help to increase the number of T cells. Lowering the amount of HIV-1 in the blood lowers the chance of infections that happen when your immune system is weak (called opportunistic infections). Tenofovir does not cure HIV-1 infection or AIDS.
You do not have to have HIV-1 infection to be treated with this medicine for HBV and vice versa.
Tenofovir does not reduce the risk of passing HIV-1 or HBV to others through sexual contact or blood contamination. Continue to practice safe sex and do not use or share dirty needles.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed this medicine for another reason.
This medicine is available only with a doctor's prescription.
This medicine is not addictive.
This medicine should not be used in children less than 12 years of age.
Before you take this medicine
When you must not take it
Do not take this medicine if you have an allergy to:
- tenofovir
- or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue, throat or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you are already taking a combination tablet that contains tenofovir or you are already taking adefovir.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- kidney problems
- bone problems
- liver problems including HBV
- HIV-1 infection
Tell your doctor if you are pregnant or plan to become pregnant. The effects of this medicine on pregnant women or their unborn babies are not known.
Tell your doctor if you are breastfeeding. Tenofovir has been found in breast milk at low concentrations and may affect your baby.
Do not breastfeed if you have HIV-1 or HBV. If you are a woman who has or will have a baby, talk with your doctor or pharmacist about the best way to feed your baby. If your baby does not already have HIV-1 or HBV, there is a chance that the baby can get HIV-1 or HBV through breastfeeding.
If you have not told your doctor about any of the above, tell them before you start taking this medicine.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interact with tenofovir. These include:
- didanosine
- atazanavir
- lopinavir/ritonavir
- ledipasvir/sofosbuvir
- sofosbuvir/velpatasvir
If you are taking any of these you may need a different dose or you may need to take different medicines. You may need to be followed more carefully if you are taking these medicines together.
Other medicines not listed above may also interact with tenofovir.
How to take this medicine
Follow carefully all directions given to you by your doctor. Their instructions may be different to the information in this leaflet.
- If you are taking this medicine to treat HIV-1 or if you have HIV-1 and HBV co-infection and are taking this medicine, always take this medicine in combination with other anti-HIV-1 medicines.
This medicine and other similar medicines, may be less likely to work in the future if you are not taking tenofovir disoproxil fumarate with other anti-HIV-1 medicines because you may develop resistance to those medicines. If you have any questions about what medicines you should or should not be taking, please see your doctor or pharmacist. - If you have been given this medicine to treat CHB, you are advised to get a HIV-1 test before you start taking this medicine and at any time after that when there is a chance you were exposed to HIV-1.
How much to take
Your doctor will tell you how much of this medicine you should take, depending on your condition and whether you are taking any other medicines.
The usual dose is one tablet once a day. If you have kidney problems, your doctor may recommend that you take this medicine less frequently.
How to take it
Swallow the tablet whole with water.
When to take it
Take this medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It is best taken with a meal or just afterwards.
How long to take it for
Continue taking your medicine for as long as your doctor tells you.
Make sure you always have enough supply of tenofovir. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to tenofovir and become harder to treat. If you are taking this medicine to treat CHB, stopping treatment may result in very severe hepatitis and serious liver problem.
If you forget to take it
It is important that you do not miss any doses.
If it is almost time to take your next dose, skip the missed dose and take your next dose at the usual time.
Otherwise, take it as soon as you remember and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for missed doses. This may increase the chance of you experiencing side effects.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice or go to the Emergency department at your nearest hospital if you think that you or anyone else may have taken too much of this medicine. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking this medicine
Things you must do
If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking this medicine.
Tell any other doctors, dentists and pharmacists who are treating you that you take this medicine.
Tell your doctor if you plan to have any vaccinations.
If you become pregnant or plan to breastfeed while taking this medicine, tell your doctor immediately.
Keep all your doctor's appointments so that your progress can be checked. Your doctor may occasionally do tests to make sure the medicine is working and to prevent side effects.
Things you must not do
Do not breastfeed if you are taking tenofovir.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not take your medicine to treat any other complaint unless your doctor tells you to.
Do not stop taking your medicine, or change the dosage, without first checking with your doctor. It is extremely important that you do not stop taking this medicine without your doctor's advice. If you have a Hepatitis B infection, you may have a 'flare-up', where the disease suddenly returns in a worse way than before. This flare-up may lead to liver failure and possibly liver transplantation or death.
After stopping this medicine, tell your doctor immediately about any new, unusual, or worsening symptoms that you notice after stopping treatment. After you stop taking this medicine, your doctor will still need to check your health and take blood tests to check your liver for several months.
Things to be careful of
Some patients taking this medicine have experienced dizziness. Make sure you know how you react to this medicine before you drive a car, operate machinery or do anything else that could be dangerous if you are dizzy.
Side effects
Tell your doctor and pharmacist as soon as possible if you do not feel well while you are taking tenofovir.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Clinical studies in patients with HIV-1:
The most common side effects are:
- diarrhoea
- nausea
- vomiting,
- dizziness.
Less common side effects include:
- flatulence (intestinal gas).
Clinical studies in patients with CHB:
The only common side effects are:
- nausea
Marketing experience:
Other side effects reported since this medicine has been marketed include:
- low blood phosphate and potassium
- shortness of breath
- increased liver enzymes, increased amylase
- inflammation of the liver
- abdominal pain
- inflammation of the pancreas
- rash
- weakness.
Some patients treated with this medicine have had kidney problems. If you have had kidney problems in the past or need to take another drug that can cause kidney problems, your doctor may need to perform additional blood tests. Kidney problems may be associated with muscle problems and softening of the bones.
Laboratory tests show changes in the bones of patients treated with this medicine. It is not known whether long-term use of this medicine will cause damage to your bones. If you have had bone problems in the past, your doctor may need to perform additional tests or may suggest additional medication.
Some patients taking antiviral drugs like this medicine have developed a condition called lactic acidosis. This is a build-up in the blood of lactic acid, the same substance that causes your muscles to burn during heavy exercise. Symptoms of lactic acidosis include nausea, vomiting, unusual or unexpected stomach discomfort, and weakness. If you notice these symptoms or if your medical condition changes suddenly, call your doctor right away.
There have been other side effects in patients taking this medicine. However, these side effects may have been due to other medicines that patients were taking or to the illness itself. Some of these side effects can be serious.
Allergic reactions
If you think you are having an allergic reaction to tenofovir disoproxil fumarate, do not take any more of this medicine and tell your doctor immediately or go to the Emergency department at your nearest hospital.
Symptoms of an allergic reaction may include some or all the following:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue, throat or other parts of the body
- rash, itching or hives on the skin
Other side effects not listed above may occur in some patients.
Storage and disposal
Storage
Keep your medicine in its pack until it is time to take it. If you take your medicine out of its pack it may not keep well.
Keep your medicine in a cool dry place where the temperature will stay below 25°C.
Do not store your medicine, or any other medicine, in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine left over.
Product description
What it looks like
300 mg tablets: light blue, almond shaped, biconvex, coated tablet. Engraved "TEN" over "300" on one side, "APO" on the other side.
Blister and bottle packs of 30 tablets
Bottle: AUST R 247315
Blister: AUST R 247314
Ingredients
Each tablet contains 300 mg tenofovir disoproxil fumarate as the active ingredient.
It also contains the following:
- lactose
- crospovidone
- calcium stearate
- colloidal anhydrous silica
- hypromellose
- macrogol 8000
- hyprolose
- titanium dioxide
- indigo carmine aluminium lake
This medicine does not contain gluten, sucrose, tartrazine or any other azo dyes.
* Not all pack types may be available.
Sponsor
Arrotex Pharmaceuticals Pty Ltd
15-17 Chapel Street
Cremorne VIC 3121
This leaflet was prepared in August 2023.
Published by MIMS September 2023
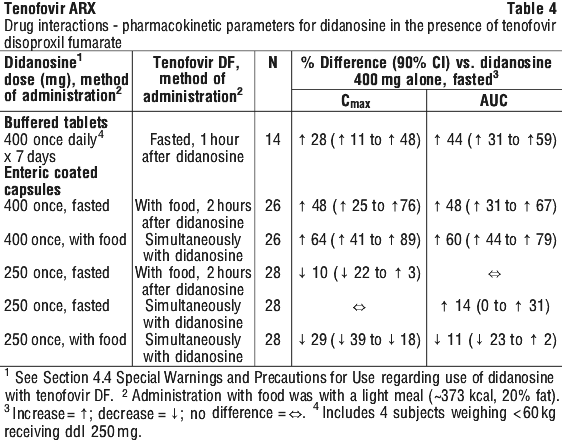
 The pharmacokinetics of tenofovir have not been evaluated in non-haemodialysis patients with creatinine clearance < 10 mL/min; therefore, no dosing recommendation is available for these patients.
The pharmacokinetics of tenofovir have not been evaluated in non-haemodialysis patients with creatinine clearance < 10 mL/min; therefore, no dosing recommendation is available for these patients. Following multiple dosing to HIV- and HBV-negative subjects receiving either chronic methadone maintenance therapy or oral contraceptives, steady state tenofovir pharmacokinetics were similar to those observed in previous studies, indicating lack of clinically significant drug interactions between these agents and tenofovir disoproxil fumarate. In a study conducted in healthy volunteers dosed with a single 600 mg dose of ribavirin, no clinically significant drug interactions were observed between tenofovir disoproxil fumarate and ribavirin.
Following multiple dosing to HIV- and HBV-negative subjects receiving either chronic methadone maintenance therapy or oral contraceptives, steady state tenofovir pharmacokinetics were similar to those observed in previous studies, indicating lack of clinically significant drug interactions between these agents and tenofovir disoproxil fumarate. In a study conducted in healthy volunteers dosed with a single 600 mg dose of ribavirin, no clinically significant drug interactions were observed between tenofovir disoproxil fumarate and ribavirin.







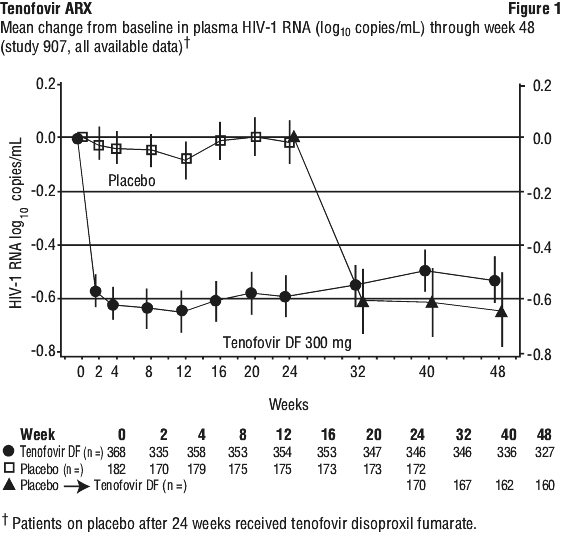 The percent of patients with HIV-1 RNA < 400 copies/mL and outcomes of patients through 48 weeks are summarized in Table 13.
The percent of patients with HIV-1 RNA < 400 copies/mL and outcomes of patients through 48 weeks are summarized in Table 13. At 24 weeks of therapy, there was a higher proportion of patients in the tenofovir disoproxil fumarate arm compared to the placebo arm with HIV-1 RNA < 50 copies/mL (19% and 1%, respectively). Mean change in absolute CD4 counts by Week 24 was +11 cells/mm3 for the tenofovir disoproxil fumarate group and -5 cells/mm3 for the placebo group. Mean change in absolute CD4 counts by Week 48 was +4 cells/mm3 for the tenofovir disoproxil fumarate group.
At 24 weeks of therapy, there was a higher proportion of patients in the tenofovir disoproxil fumarate arm compared to the placebo arm with HIV-1 RNA < 50 copies/mL (19% and 1%, respectively). Mean change in absolute CD4 counts by Week 24 was +11 cells/mm3 for the tenofovir disoproxil fumarate group and -5 cells/mm3 for the placebo group. Mean change in absolute CD4 counts by Week 48 was +4 cells/mm3 for the tenofovir disoproxil fumarate group. Achievement of plasma HIV-1 RNA concentrations of less than 400 copies/mL at Week 144 was similar between the two treatment groups for the population stratified at baseline on the basis of HIV-1 RNA concentration (≤ or > 100,000 copies/mL) and CD4 cell count (< or ≥ 200 cells/mm3). Through 144 weeks of therapy, 62% and 58% of patients in the tenofovir disoproxil fumarate and d4T arms, respectively, achieved and maintained confirmed HIV-1 RNA < 50 copies/mL. The mean increase from baseline in CD4 cell count was 263 cells/mm3 for the tenofovir disoproxil fumarate arm and 283 cells/mm3 for the d4T arm.
Achievement of plasma HIV-1 RNA concentrations of less than 400 copies/mL at Week 144 was similar between the two treatment groups for the population stratified at baseline on the basis of HIV-1 RNA concentration (≤ or > 100,000 copies/mL) and CD4 cell count (< or ≥ 200 cells/mm3). Through 144 weeks of therapy, 62% and 58% of patients in the tenofovir disoproxil fumarate and d4T arms, respectively, achieved and maintained confirmed HIV-1 RNA < 50 copies/mL. The mean increase from baseline in CD4 cell count was 263 cells/mm3 for the tenofovir disoproxil fumarate arm and 283 cells/mm3 for the d4T arm.
 In this study, tenofovir disoproxil fumarate + emtricitabine in combination with efavirenz was statistically significantly superior to lamivudine/zidovudine in combination with efavirenz with regard to the primary and secondary endpoints: achieving and maintaining HIV-1 RNA < 400 copies/mL through 48 and 144 weeks (Table 15). The difference in the proportions of responders between the tenofovir disoproxil fumarate + emtricitabine group and the lamivudine/zidovudine group was 11.4%, and the 95% CI was 4.3-18.6% (p = 0.002) at Week 48 and a difference of 12.9% (95% CI was 4.2-21.6%, p = 0.004) at Week 144.
In this study, tenofovir disoproxil fumarate + emtricitabine in combination with efavirenz was statistically significantly superior to lamivudine/zidovudine in combination with efavirenz with regard to the primary and secondary endpoints: achieving and maintaining HIV-1 RNA < 400 copies/mL through 48 and 144 weeks (Table 15). The difference in the proportions of responders between the tenofovir disoproxil fumarate + emtricitabine group and the lamivudine/zidovudine group was 11.4%, and the 95% CI was 4.3-18.6% (p = 0.002) at Week 48 and a difference of 12.9% (95% CI was 4.2-21.6%, p = 0.004) at Week 144. In the protocol defined analyses, virologic response to tenofovir disoproxil fumarate was not reduced in patients with HIV that expressed the lamivudine/abacavir-associated M184V mutation. In the absence of zidovudine-associated mutations, patients with the M184V mutation receiving tenofovir disoproxil fumarate showed a -0.84 log10 copies/mL decrease in their HIV RNA relative to placebo. In the presence of zidovudine-associated mutations, the M184V mutation did not affect the mean HIV RNA responses to tenofovir disoproxil fumarate treatment. HIV-1 RNA responses among these patients were durable through Week 48.
In the protocol defined analyses, virologic response to tenofovir disoproxil fumarate was not reduced in patients with HIV that expressed the lamivudine/abacavir-associated M184V mutation. In the absence of zidovudine-associated mutations, patients with the M184V mutation receiving tenofovir disoproxil fumarate showed a -0.84 log10 copies/mL decrease in their HIV RNA relative to placebo. In the presence of zidovudine-associated mutations, the M184V mutation did not affect the mean HIV RNA responses to tenofovir disoproxil fumarate treatment. HIV-1 RNA responses among these patients were durable through Week 48.
 Tenofovir disoproxil fumarate was associated with statistically significantly greater proportions of patients with undetectable HBV DNA (< 169 copies/mL [< 29 IU/mL]; the limit of quantification of the Roche COBAS TaqMan HBV assay), when compared with adefovir dipivoxil (Study 0102; 91% and 56%, respectively and Study 0103; 69% and 9%, respectively).
Tenofovir disoproxil fumarate was associated with statistically significantly greater proportions of patients with undetectable HBV DNA (< 169 copies/mL [< 29 IU/mL]; the limit of quantification of the Roche COBAS TaqMan HBV assay), when compared with adefovir dipivoxil (Study 0102; 91% and 56%, respectively and Study 0103; 69% and 9%, respectively).
 Paired baseline and Week 240 liver biopsy data were available for 331/489 patients who remained in Studies 0102 and 0103 (see Table 21). Ninety-five percent (225/237) of patients without cirrhosis at baseline and 99% (93/94) of patients with cirrhosis at baseline had either no change or an improvement in fibrosis (Ishak fibrosis score). Of the 94 patients with cirrhosis at baseline (Ishak fibrosis score 5-6), 26% (24) experienced no change in Ishak fibrosis score and 72% (68) experienced reversal of cirrhosis by Week 240 with a reduction in Ishak fibrosis score of at least 2 points except for one patient with an initial Ishak score of five.
Paired baseline and Week 240 liver biopsy data were available for 331/489 patients who remained in Studies 0102 and 0103 (see Table 21). Ninety-five percent (225/237) of patients without cirrhosis at baseline and 99% (93/94) of patients with cirrhosis at baseline had either no change or an improvement in fibrosis (Ishak fibrosis score). Of the 94 patients with cirrhosis at baseline (Ishak fibrosis score 5-6), 26% (24) experienced no change in Ishak fibrosis score and 72% (68) experienced reversal of cirrhosis by Week 240 with a reduction in Ishak fibrosis score of at least 2 points except for one patient with an initial Ishak score of five. When the data were evaluated including only patients that completed 384 weeks of therapy (observed (missing data is excluded) and data after the addition of emtricitabine included; on therapy analysis), in the group of patients who received 48 weeks of double-blind treatment with tenofovir disoproxil fumarate followed by open-label treatment with tenofovir disoproxil fumarate; 99% (173/174) and 100% (88/88) of patients had HBV DNA < 400 copies/mL and 88% (141/160) and 81% (70/86) of patients had ALT normalization at Week 384, in Studies 0102 and 0103 respectively. In Study 0103, HBeAg loss was reported for 44% (31/70) of patients and 28% (19/68) of patients experienced HBeAg seroconversion. 14% of patients experienced HBsAg loss and 12% of patients experienced HBsAg seroconversion by Week 384. In study 102, HBsAg loss and seroconversion were 1% in both treatment groups.
When the data were evaluated including only patients that completed 384 weeks of therapy (observed (missing data is excluded) and data after the addition of emtricitabine included; on therapy analysis), in the group of patients who received 48 weeks of double-blind treatment with tenofovir disoproxil fumarate followed by open-label treatment with tenofovir disoproxil fumarate; 99% (173/174) and 100% (88/88) of patients had HBV DNA < 400 copies/mL and 88% (141/160) and 81% (70/86) of patients had ALT normalization at Week 384, in Studies 0102 and 0103 respectively. In Study 0103, HBeAg loss was reported for 44% (31/70) of patients and 28% (19/68) of patients experienced HBeAg seroconversion. 14% of patients experienced HBsAg loss and 12% of patients experienced HBsAg seroconversion by Week 384. In study 102, HBsAg loss and seroconversion were 1% in both treatment groups.


 Tenofovir is efficiently removed by haemodialysis with an extraction coefficient of approximately 54%. Following a single 300 mg dose of tenofovir disoproxil fumarate, a four-hour haemodialysis session removed approximately 10% of the administered tenofovir dose.
Tenofovir is efficiently removed by haemodialysis with an extraction coefficient of approximately 54%. Following a single 300 mg dose of tenofovir disoproxil fumarate, a four-hour haemodialysis session removed approximately 10% of the administered tenofovir dose. Chemical name: 9-[(R)-2[[bis[[(isopropoxycarbonyl) oxy]methoxy]-phosphinyl]- methoxy] propyl]adenine fumarate (1:1).
Chemical name: 9-[(R)-2[[bis[[(isopropoxycarbonyl) oxy]methoxy]-phosphinyl]- methoxy] propyl]adenine fumarate (1:1).