What is in this leaflet
Read all of this leaflet carefully before you start taking this medicine.
This leaflet answers some common questions about TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist about your medical condition or treatment. If you have further questions, please ask your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is used for
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is used to treat Human Immunodeficiency Virus (HIV) infection in adults. This medicine must be taken in combination with other anti-HIV medicines. TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 consists of two medicines:
- tenofovir disoproxil maleate
- emtricitabine
These are combined in one tablet to help control Human Immunodeficiency Virus (HIV) infection.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 contains two active ingredients that belong to a group of antiviral medicines known as nucleoside and nucleotide reverse transcriptase inhibitors (NRTI).
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is used:
- to treat Human Immunodeficiency Virus-1 (HIV-1) infection in adults when taken in combination with other anti-HIV medicines;
- to help reduce the risk of getting HIV infection when used with safer sex practices in:
HIV-negative men who have sex with men, who are at high risk of getting infected with HIV-1 through sex.
Male-female sex partners when one partner has HIV-1 infection and the other does not.
When TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is used to treat HIV infection
When used with other HIV-1 medicines to treat HIV-1 infection, TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 may help:
- Reduce the amount of HIV-1 in your blood. This is called "viral load".
- Increase the number of CD4+ (T) cells in your blood that help fight off other infections.
Reducing the amount of HIV-1 and increasing the CD4+ (T) cells in your blood may help improve your immune system.
This may reduce your risk of death or infections that can happen when your immune system is weak.
Use in Children and Elderly
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is for adults.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are under the age of 18 years.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are over the age of 65 before discussing with your doctor.
Does TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 cure HIV OR AIDS?
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is not a cure for HIV infection or AIDS. While taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 you may still develop infections or other illnesses associated with HIV infection.
If you have HIV-1 infections, you must keep taking HIV-1 medicines to control HIV-1 infection and decrease HIV-related illnesses.
Does TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 reduce the risk of HIV to others?
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 does not reduce the risk of passing HIV to others through sexual contact or blood contamination. It is important to continue to take appropriate precautions to prevent passing HIV to others.
When TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is used to reduce the risk of HIV infection
When used with safer sex practices, TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 may help to reduce the risk of getting HIV-1 infection if you are at high risk of getting HIV infection.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 reduces the risk of getting HIV-1 when you have been taking it before you are exposed to HIV-1.
Before you take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200
Who must not take it
Together with your doctor, you need to decide whether TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is right for you.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are allergic to:
- tenofovir,
- tenofovir disoproxil maleate,
- tenofovir disoproxil fumarate,
- any of the other ingredients of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- rash, itching or hives on the skin
- swelling of the face, lips, tongue or other parts of the body
The ingredients of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 are listed in the product description section of this leaflet.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are already taking any of the components of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 (tenofovir disoproxil maleate or emtricitabine).
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are taking lamivudine.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are taking adefovir dipivoxil.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are taking tenofovir alafenamide.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 after the expiry or "use by" date (EXP) printed on the packaging. If you take it after the expiry date has passed, it may not work as well.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if the packaging is torn or shows signs of tampering.
If you are not sure whether you should be taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200, talk to your doctor.
For people using TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce the risk of getting HIV-1 infection:
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 can only help reduce your risk of getting HIV-1 before you are infected.
Do not take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to help reduce your risk of getting HIV-1 if:
- you already have HIV-1 infection. If you are HIV-positive, you need to take other medicines with TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to treat HIV. TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 by itself is not a complete treatment for HIV.
- you do not know your HIV-1 infection status. You may already be HIV-positive. You need to take other HIV-1 medicines with TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to treat HIV-1.
- Many HIV-1 tests can miss HIV-1 infection in a person who has recently become infected. If you have flu-like symptoms, you could have recently become infected with HIV-1. Tell your healthcare provider if you had a flu-like illness within the last month before starting TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 or at any time while taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. Symptoms of new HIV-1 infection include: tiredness, fever, joint or muscle aches, headache, sore throat, vomiting or diarrhoea, rash, night sweats or enlarged lymph nodes in the neck or groin.
Before you start to take it:
Tell your doctor if you are allergic to foods, dyes, preservatives or any other medicines.
Tell your doctor if you are pregnant, trying to become pregnant or breast feeding.
The safe use of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 in pregnancy has not been demonstrated. For this reason, it is important that women of child-bearing age receiving treatment with TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 use an effective method of contraception to avoid becoming pregnant.
If you are a female who is taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce the risk of getting HIV-1 infection and you become pregnant while taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200, talk to your healthcare provider to decide if you should keep taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200.
The active substances in this medicine (tenofovir disoproxil maleate and emtricitabine) have been found in breast milk at low concentrations.
Consequently, it is recommended that nursing mothers do not breast-feed during treatment with TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. In general, women infected with HIV should not breast-feed their infants in order to avoid transmission of HIV to their newborn infant.
Tell your doctor if you have liver problems, including hepatitis B, or C virus infection.
Tell your doctor if you are taking medication to treat your hepatitis C virus (HCV) infection (e.g. ledipasvir/sofosbuvir, sofosbuvir/velpatasvir.
Tell your doctor if you have kidney problems.
Tell your doctor if you have or have ever had abnormal bones or bone difficulties.
This medicine is only available from a pharmacist after it has been prescribed by a doctor who specialises in the treatment of HIV infection.
If you wish to continue receiving treatment with TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 it is important you remain under the care of a hospital or doctor who specialises in the treatment of HIV infection.
Avoid doing things that increase your risk of getting HIV-1 or spreading HIV-1 to other people:
- Do not have any kind of sex without protection. Always practice safer sex. Use latex or non-latex condoms, except lambskin, to reduce contact with semen, vaginal fluids, or blood.
- Do not share personal items that can have blood or body fluids on them, such as toothbrushes and razor blades.
- Do not share or re-use needles or other injection equipment.
Ask your healthcare provider if you have any questions about how to prevent getting HIV-1 or spreading HIV-1 to other people.
If you have a long standing viral infection of your liver (hepatitis B) it may flare up when you stop taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. This can cause serious illness particularly if your liver is already not working very well. If you have both HIV and hepatitis B, when you start taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 and even after you stop, your doctor is likely to arrange tests from time to time to check how well your liver is working.
Taking other medicines
If you have HIV infections your doctor will generally prescribe TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 in combination with other anti-HIV medicines.
Tell your doctor if you are taking any other medicines, including medicines you buy without a prescription from a pharmacy, supermarket or health food shop.
Tell your doctor if are taking:
- didanosine (also known as ddI or Videx).
- ledipasvir with sofosbuvir (HARVONI®)
- sofosbuvir/velpatasvir (EPCLUSA®)
- sofosbuvir/velpatasvir/voxilaprevir (e.g. VOSEVI®)
Some medicines may affect the way others work. Your doctor or pharmacist will be able to tell you what to do when taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets with other medicines.
How to take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200
Take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 exactly as prescribed. The usual dose is one TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablet once daily.
Take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 at the same time each day to keep TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 blood levels constant.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is best taken with a meal or just afterwards, however taking it without food should not reduce the effectiveness of the medicine.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is absorbed rapidly. Do not take another TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 dose if vomiting has occurred unless it occurs within 1 hour after taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200.
How to take it
Take one TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablet once daily or as advised by your doctor.
If you are not sure how much TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 you should take, check with your doctor or pharmacist. Do not change the amount of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 you take unless told to do so by your doctor.
Your doctor will tell you how much TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to take and how often to take it. You will also find this information on the label of your medicine container.
Because your medicine helps to control your condition, but does not cure it, you will need to take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 every day. If you are taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce the risk of HIV-1 infection, take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 every day for the period of time as prescribed by your doctor.
Do not miss any doses of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. Missing a dose lowers the amount of medicine in your blood.
Do not stop taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 without first talking to your doctor.
If you forget to take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200
It is important to take the prescribed daily dose in order to get the maximum benefit of treatment.
If you forget to take your daily dose of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 take it as soon as you remember that day and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for a forgotten dose.
Do not take more than one TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablet in a day.
If you take too much (overdose)?
Immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26), or in New Zealand the Poisons Centre (telephone 0800 POISON or 0800 764 766) or go to the accident and emergency department at your nearest hospital if you think you or anyone else may have taken too many TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets. Do this even if there are no signs of discomfort or poisoning. This may need urgent medical attention.
While you are taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200
Things you must do
Tell your doctor or pharmacist that you are taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 if you are about to be started on any other medicines.
Tell your doctor if you become pregnant or are trying to become pregnant.
Tell your doctor if for any reason you have not taken your medicine exactly as prescribed.
If you are taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce your risk of getting HIV
Just taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 may not keep you from getting HIV.
You must continue using safer sex practices while you are taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce your risk of getting HIV.
You must stay HIV-negative to keep taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce your risk of infection.
Know your HIV status and the HIV status of your partners.
Get tested for HIV at least every 3 months or when your healthcare provider tells you.
Get tested for other sexually transmitted infections such as syphilis and gonorrhea. These infections make it easier for HIV to infect you. TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 will not stop you from getting these other infections.
If you think you were exposed to HIV, tell your healthcare provider right away. They may want to do more tests to be sure you are still HIV-negative.
Get information and support to help reduce risky sexual behaviour.
Have fewer sex partners.
Do not miss any doses of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. Missing doses may increase your risk of getting HIV infection.
If you do become HIV-positive, you need more medicine than TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 alone to treat HIV. TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 by itself is not a complete treatment for HIV.
If you have HIV and take only TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200, over time your HIV may become harder to treat.
Things you must not do
Do not stop taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 or change the dose without first checking with your doctor.
Do not give this medicine to anyone else even if their symptoms seem similar to yours.
Do not use TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to treat any other complaints unless you doctor says to.
Things to be careful of
Be careful driving or operating machinery until you know how TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 affects you.
SIDE EFFECTS
Like all medicines, TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 can have side effects, although not everybody gets them. Some may be serious and need medical attention.
Check with your doctor as soon as possible if you have any problems while taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- Diarrhoea
- Nausea
- tiredness
- Headache
- Vomiting
- Dizziness
- Depression
- Problems sleeping
- Abnormal dreams
- Rash
Common side effects in people who take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 to reduce the risk of getting HIV-1 infection include:
- stomach-area (abdomen) pain
- headache
- decreased weight
Ask your doctor or pharmacist to answer any question you may have about these or other effects.
Allergy
Some people are allergic to medicines.
If you have any of the following symptoms soon after taking your medicine, DO NOT TAKE ANY MORE TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 and tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital.
- Skin troubles such as lumpy skin rash or "hives"
- Swelling of the face, lips, mouth or throat which may cause difficulty in swallowing or breathing
- Wheezing, chest pain or tightness
- Fainting
These are very serious effects. If you have them, you may have a serious allergic reaction. You may need urgent medical attention or hospitalisation. All of these side effects are very rare.
Pancreatitis
If you have any of the following symptoms after starting your medication, tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital.
- Severe stomach pain or cramps
- Nausea
- Vomiting
These side effects may be due to a condition called pancreatitis which sometimes occurs in patients taking anti-HIV medicines.
Serious Liver Problems (hepatotoxicity)
If you have any of the following symptoms while taking your medication, tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital.
- Your skin or the white part of your eyes turns yellow (jaundice)
- Your urine turns dark
- Your bowel movements (stools) turn light in colour
- You don't feel like eating food for several days or longer
- Nausea
- Stomach pains
These side effects may be due to a condition called hepatotoxicity with liver enlargement and fat deposits in the liver (steatosis) which sometimes occurs in patients taking anti-HIV medicines.
Lactic Acidosis
If you have any of the following symptoms after taking your medication, tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital:
- You feel very weak or tired
- You have unusual (not normal) muscle pain
- You have trouble breathing
- You have stomach pain with nausea and vomiting
- You feel cold, especially in your arms and legs
- You feel dizzy or light headed
- You have a fast or irregular heartbeat
These side effects may be due to a condition called lactic acidosis (build-up of an acid in the blood).
Lactic acidosis can be a medical emergency and may need to be treated in the hospital.
You may be more likely to get lactic acidosis or liver problems if you are female, very overweight (obese), or have been taking similar nucleoside analog-containing medicines, like TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200, for a long time.
Hepatic Flares
Your doctor should test you to see if you have chronic hepatitis B infection before you start taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200.
If you have chronic hepatitis B infection you should not stop your TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 treatment without first discussing this with your doctor, as some patients have had blood tests or symptoms indicating a worsening of their hepatitis ("hepatic flare") after stopping individual components (tenofovir disoproxil maleate and emtricitabine) of TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. You may require medical exams and blood tests for several months after stopping treatment.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is not approved for the treatment of hepatitis B, so you must discuss your hepatitis B therapy with your healthcare provider.
Other possible side effects:
This list of side effects is not complete.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 may cause other serious side effects. Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list:
New and worse kidney problems
If you have had kidney problems in the past or need to take another drug that can cause kidney problems, your healthcare provider may need to perform additional blood tests to check your kidneys.
Bone problems
Bone problems can happen in some people who take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. Bone problems include bone pain, or softening or thinning of bones, which may lead to fractures. Your healthcare provider may need to do tests to check your bones.
Signs and symptoms of inflammation
In some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response, which lets the body fight infections that may have been present with no obvious symptoms. If you notice any symptoms of infection, please tell your doctor immediately.
Some people may get other side effects while taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200. If you are concerned, talk to your doctor or pharmacist.
Ask your doctor or pharmacist if you don't understand anything in this list.
Do not be alarmed by this list of possible side-effects. Most of them are very rare and you may not experience any of them.
After taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200
Storage
Keep TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets where children cannot reach them. A locked cupboard at least one-and-a half metres above the ground is a good place to store them.
Keep TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets in a cool, dry place where it stays below 25°C.
Do not store TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets or any other medicine in a bathroom or near a sink.
Do not leave TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep your TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets in the bottle with the cap tightly closed until you take them.
If you take TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets out of their pack they may not keep well.
Disposal
If your doctor tells you to stop taking TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200, or the tablets have passed their expiry date, ask your pharmacist what to do with any tablets left over.
Product description
What it looks like
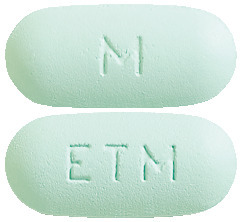
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablets are light green, film-coated, capsule shaped, biconvex tablets debossed with 'M' on one side of the tablet and 'ETM' on the other side.
Ingredients
Each TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablet contains the following active ingredients:
- 300 mg tenofovir disoproxil maleate
- 200 mg emtricitabine
Each TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 tablet contains the following inactive ingredients
- microcrystalline cellulose
- hyprolose
- iron oxide red
- colloidal anhydrous silica
- lactose monohydrate
- magnesium stearate
- Opadry II complete film coating system 32K510087 GREEN (ARTG PI No: 114524)
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS also contains lactose.
Sponsor
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 is supplied in Australia by:
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
Australian registration numbers:
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200 film coated tablet bottle:
AUST R 265834
This leaflet was prepared in August 2022.
TENOFOVIR DISOPROXIL EMTRICITABINE VIATRIS 300/200_cmi\Aug22/00
Published by MIMS October 2022


 Since tenofovir and emtricitabine are primarily eliminated by the kidneys, co-administration of combination tenofovir disoproxil/emtricitabine tablet with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of tenofovir, emtricitabine, and/or other renally eliminated drugs.
Since tenofovir and emtricitabine are primarily eliminated by the kidneys, co-administration of combination tenofovir disoproxil/emtricitabine tablet with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of tenofovir, emtricitabine, and/or other renally eliminated drugs.









 Tenofovir and emtricitabine belong to the nucleoside and nucleotide reverse transcriptase inhibitors pharmacotherapeutic group (ATC code: J05AF30).
Tenofovir and emtricitabine belong to the nucleoside and nucleotide reverse transcriptase inhibitors pharmacotherapeutic group (ATC code: J05AF30). In this study, tenofovir disoproxil tablet + emtricitabine tablet in combination with efavirenz was statistically significantly superior to lamivudine/zidovudine in combination with efavirenz with regards to the primary and secondary endpoints: achieving and maintaining HIV-1 RNA < 400 copies/mL through 48 and 144 weeks (Table 15). The difference in the proportions of responders between the tenofovir disoproxil tablet + emtricitabine tablet group and the Combivir group was 11.4%, and the 95% CI was 4.3% to 18.6% (p=0.002) at week 48 and a difference of 12.9% (95% CI was 4.2% to 21.6%, p=0.004) at week 144.
In this study, tenofovir disoproxil tablet + emtricitabine tablet in combination with efavirenz was statistically significantly superior to lamivudine/zidovudine in combination with efavirenz with regards to the primary and secondary endpoints: achieving and maintaining HIV-1 RNA < 400 copies/mL through 48 and 144 weeks (Table 15). The difference in the proportions of responders between the tenofovir disoproxil tablet + emtricitabine tablet group and the Combivir group was 11.4%, and the 95% CI was 4.3% to 18.6% (p=0.002) at week 48 and a difference of 12.9% (95% CI was 4.2% to 21.6%, p=0.004) at week 144. The percent of patients with HIV RNA < 400 copies/mL and outcomes of patients through 48 weeks are summarised in Table 16.
The percent of patients with HIV RNA < 400 copies/mL and outcomes of patients through 48 weeks are summarised in Table 16. At 24 weeks of therapy, there was a higher proportion of patients in the tenofovir disoproxil arm compared to the placebo arm with HIV RNA < 50 copies/mL (19% and 1%, respectively). Mean change in absolute CD4 counts by week 24 was +12 cells/mm3 for the tenofovir group and -5 cells/mm3 for the placebo group. Mean change in absolute CD4 counts by week 48 was +4 cells/mm3 for the tenofovir disoproxil group.
At 24 weeks of therapy, there was a higher proportion of patients in the tenofovir disoproxil arm compared to the placebo arm with HIV RNA < 50 copies/mL (19% and 1%, respectively). Mean change in absolute CD4 counts by week 24 was +12 cells/mm3 for the tenofovir group and -5 cells/mm3 for the placebo group. Mean change in absolute CD4 counts by week 48 was +4 cells/mm3 for the tenofovir disoproxil group. Achievement of plasma HIV-1 RNA concentrations of less than 400 copies/mL at week 144 was similar between the two treatment groups for the population stratified at baseline on the basis of HIV-1 RNA concentration (≤ or > 100,000 copies/mL) and CD4 cell count (< or ≥ 200 cells/mm3). Through 144 weeks of therapy, 62% and 58% of patients in the tenofovir disoproxil and d4T arms, respectively achieved and maintained confirmed HIV-1 RNA < 50 copies/mL. The mean increase from baseline in CD4 cell count was 263 cells/mm3 for the tenofovir disoproxil arm and 283 cells/mm3 for the d4T arm.
Achievement of plasma HIV-1 RNA concentrations of less than 400 copies/mL at week 144 was similar between the two treatment groups for the population stratified at baseline on the basis of HIV-1 RNA concentration (≤ or > 100,000 copies/mL) and CD4 cell count (< or ≥ 200 cells/mm3). Through 144 weeks of therapy, 62% and 58% of patients in the tenofovir disoproxil and d4T arms, respectively achieved and maintained confirmed HIV-1 RNA < 50 copies/mL. The mean increase from baseline in CD4 cell count was 263 cells/mm3 for the tenofovir disoproxil arm and 283 cells/mm3 for the d4T arm.
 In the protocol defined analyses, virologic response to tenofovir disoproxil was not reduced in patients with HIV that expressed the lamivudine/ abacavir-associated M184V mutation. In the absence of zidovudine-associated mutations, patients with the M184V mutation receiving tenofovir disoproxil showed a -0.84 log10 copies/mL decrease in their HIV RNA relative to placebo. In the presence of zidovudine-associated mutations, the M184V mutation did not affect the mean HIV RNA responses to tenofovir disoproxil treatment. HIV-1 RNA responses among these patients were durable through week 48.
In the protocol defined analyses, virologic response to tenofovir disoproxil was not reduced in patients with HIV that expressed the lamivudine/ abacavir-associated M184V mutation. In the absence of zidovudine-associated mutations, patients with the M184V mutation receiving tenofovir disoproxil showed a -0.84 log10 copies/mL decrease in their HIV RNA relative to placebo. In the presence of zidovudine-associated mutations, the M184V mutation did not affect the mean HIV RNA responses to tenofovir disoproxil treatment. HIV-1 RNA responses among these patients were durable through week 48.
 The mean increase from baseline in CD4 cell count was 29 cells/mm3 for the emtricitabine arm and 61 cells/mm3 for the lamivudine arm.
The mean increase from baseline in CD4 cell count was 29 cells/mm3 for the emtricitabine arm and 61 cells/mm3 for the lamivudine arm. The mean increase from baseline in CD4 cell count was 168 cells/mm3 for the emtricitabine arm and 134 cells/mm3 for the d4T arm.
The mean increase from baseline in CD4 cell count was 168 cells/mm3 for the emtricitabine arm and 134 cells/mm3 for the d4T arm.
 Molecular formula: C19H30N5O10P.C4H4O4. Molecular weight: 635.51.
Molecular formula: C19H30N5O10P.C4H4O4. Molecular weight: 635.51. Molecular formula: C8H10FN3O3S. Molecular weight: 247.24.
Molecular formula: C8H10FN3O3S. Molecular weight: 247.24.