1 Name of Medicine
Teriparatide (rbe).
Terrosa solution for injection is a biosimilar medicine to Forteo solution for injection. The evidence for comparability supports the use of Terrosa for the listed indications.
2 Qualitative and Quantitative Composition
The active ingredient for Terrosa solution for injection is teriparatide.
Terrosa solution for injection contains 250 microgram/mL of teriparatide.
Teriparatide (rbe) is produced in E. coli, using recombinant DNA technology, is identical to the 34-N-terminal amino acid sequence of endogenous human parathyroid hormone.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
Terrosa is a sterile, colourless, clear solution for injection with a pH of 3.8 - 4.5.
4.1 Therapeutic Indications
Terrosa is indicated for the treatment of osteoporosis in postmenopausal women and the treatment of primary osteoporosis in men when other agents are considered unsuitable and when there is a high risk of fractures.
Terrosa is indicated for the treatment of osteoporosis associated with sustained systemic glucocorticoid therapy in women and men at high risk for fracture.
4.2 Dose and Method of Administration
Dosage.
The recommended dose of Terrosa is 20 microgram administered once daily by subcutaneous injection in the thigh or abdomen.
Based on clinical experience, treatment with teriparatide is recommended for a lifetime duration of 24 months treatment (for post-treatment efficacy, see Section 5.1 Pharmacodynamic Properties, Clinical trials). Terrosa should be prescribed to patients with a full explanation and their informed consent on the lifetime duration of 24 months treatment.
Following cessation of teriparatide therapy, patients may be continued on other osteoporosis therapies.
Calcium and vitamin D supplements are advised in patients with a low dietary intake of these nutrients.
Use in males.
Primary or secondary hypogonadism should first be excluded and, if relevant, be treated (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Method of administration.
Terrosa is a clear and colourless solution for injection. Terrosa should not be used if the solution is cloudy, coloured or contains visible particles.
Terrosa pre-filled pen delivers 20 microgram of teriparatide per dose. Each pre-filled pen contains dosing for 28 treatment days. Before administration patient must be educated to use the proper injection techniques. Please refer to the Instructions for Use within the Consumer Medicine Information.
No needles are supplied with the Terrosa pre-filled pen. Patient must purchase Becton Dickson or Ypsomed's 29-31 gauge needles from their pharmacy. A new sterile needle must be used for every injection.
To prevent the possible transmission of disease, each pre-filled pen must be used by one patient only, even if the needle is changed.
Batch (Lot) number of each pre-filled pen and the date of first injection should be recorded by the patient on a calendar. Patients must be reminded to retain their Terrosa pre-filled pen for the total duration of their 28 treatment days.
Data are not available on the safety or efficacy of intravenous or intramuscular injection of teriparatide.4.3 Contraindications
Terrosa should not be given to patients with hypersensitivity to teriparatide or to any of its excipients.
Pregnancy and breast-feeding (see Section 4.6 Fertility, Pregnancy and Lactation).
Pre-existing hypercalcaemia.
Severe renal impairment.
Metabolic bone diseases (including hyperparathyroidism and Paget's disease of the bone) other than primary osteoporosis or glucocorticoid-induced osteoporosis.
Unexplained elevations of alkaline phosphatase.
Prior external beam or implant radiation therapy to the skeleton.
Patients with skeletal malignancies or bone metastases should be excluded from treatment with teriparatide.
4.4 Special Warnings and Precautions for Use
Duration of treatment.
The maximum lifetime exposure to teriparatide for an individual patient is 24 months (see Section 4.2 Dose and Method of Administration). For post-treatment efficacy (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Risk of osteosarcoma.
In male and female rats, teriparatide caused an increase in the incidence of osteosarcoma (a malignant bone tumour) that was dependent on dose and treatment duration. The effect was observed at systemic exposures to teriparatide ranging from 3 to 60 times the exposure in humans given a 20 microgram dose.
However, no increased risk was identified in a study in which 30 monkeys were treated with teriparatide for 18 months and then observed for a further 3 years. Postmarketing data in humans has not identified an increased risk.
To minimise the potential risk of osteosarcoma (seen in the life-time rat studies):
1. The maximum lifetime exposure to teriparatide for an individual patient is 24 months (see Section 4.2 Dose and Method of Administration; Section 5.1 Pharmacodynamic Properties).
2. Teriparatide should not be prescribed to patients where there is an increased background risk of osteosarcoma (such as Paget's disease of bone, prior radiation therapy involving the skeleton, open epiphysis, unexplained elevations of alkaline phosphatase) (see Section 4.3 Contraindications; Section 5.3 Preclinical Safety Data, Carcinogenicity).
Terrosa should be prescribed to patients with a full explanation and their informed consent on the lifetime duration of 24 months treatment.
Information for patients.
For safe and effective use of Terrosa, the physician should inform the patient on the following:
General.
Patients will need to read the Consumer Medicine Information leaflet for the pre-filled pen Instruction for Use before starting therapy with Terrosa and re-read them each time the prescription is renewed.
Osteosarcoma in rats.
Patients should be made aware that teriparatide caused osteosarcoma in rats and that the clinical relevance of these findings is unknown.
Consent form.
Use of teriparatide is restricted to 24 months lifetime duration.
Hypercalcaemia.
Teriparatide has not been studied in patients with pre-existing hypercalcaemia. Patients with pre-existing hypercalcaemia should be excluded from treatment with teriparatide because of the possibility of exacerbating hypercalcaemia (see Section 4.3 Contraindications).
In normocalcaemic patients, slight and transient elevations of serum calcium concentrations have been observed following teriparatide injection. Serum calcium concentrations reach a maximum between 4 and 6 hours and return to baseline by 16 to 24 hours after each dose of teriparatide. Routine calcium monitoring during therapy is not required.
Bone disorders other than osteoporosis.
Patients with metabolic bone diseases other than primary osteoporosis (including hyperparathyroidism and Paget's disease of the bone (see Section 4.3 Contraindications) and those with otherwise unexplained elevations of alkaline phosphatase should generally be excluded from treatment with teriparatide. Patients with skeletal malignancies or bone metastases should also be excluded from treatment with teriparatide.
Urolithiasis.
Teriparatide has not been studied in patients with active urolithiasis. Teriparatide should be used with caution in patients with active or recent urolithiasis because of the potential to exacerbate this condition.
Hypotension.
In short-term clinical studies with teriparatide isolated episodes of transient orthostatic hypotension were observed. Typically, an event began within 4 hours of dosing and spontaneously resolved within a few minutes to a few hours. When transient orthostatic hypotension occurred, it happened within the first several doses, was relieved by placing subjects in a reclining position, and did not preclude continued treatment.
Use in the elderly.
No differences in teriparatide pharmacokinetics were detected with regard to age (range 31 to 85 years). Dosage adjustment based on age is not required.
Paediatric use.
Teriparatide has not been studied in paediatric populations. Teriparatide should not be used in paediatric patients or young adults with open epiphyses.
Effects on laboratory tests.
Serum calcium.
Teriparatide transiently increases serum calcium, with the maximal effect observed at approximately 4 to 6 hours post-dose. By 16 hours post-dose, serum calcium generally has returned to, or near, baseline. These effects should be kept in mind because serum calcium concentrations observed within 16 hours after a dose may reflect the pharmacologic effect of teriparatide. Persistent hypercalcaemia was not observed in clinical trials with teriparatide. If persistent hypercalcaemia is detected, treatment with teriparatide should be discontinued pending further evaluation of the cause of hypercalcaemia. Patients known to have an underlying hypercalcaemic disorder, such as primary hyperparathyroidism, should not be treated with teriparatide (see Section 4.4 Special Warnings and Precautions for Use, Hypercalcaemia).
Teriparatide has not been studied in non-ambulant patients, thus monitoring of serum calcium may be appropriate when a previously ambulant patient is confined to bed.
Urinary calcium.
Teriparatide may cause small increases in urinary calcium excretion. However, in the clinical trials, the incidence of hypercalciuria in teriparatide patients did not differ from that in the placebo-treated patients.
Use in renal impairment.
No significant adverse renal effects were observed in long-term clinical studies. Assessments included creatinine clearance, measurements of blood urea nitrogen (BUN), creatinine, and electrolytes in serum, urine specific gravity and pH and examination of urine sediment. Long-term evaluation of patients with severe renal insufficiency, patients undergoing acute or chronic dialysis, or patients who have a functioning renal transplant has not been performed.
Caution should be exercised in patients with moderate renal impairment.
Contraindicated for use in severe renal impairment.
Younger adult population.
Experience in the younger adult population, including premenopausal women, is limited (see Section 5.1 Pharmacodynamic Properties). Treatment should only be initiated if the benefit clearly outweighs risks in this population.
Contraception and women of childbearing potential.
Women of childbearing potential should use effective methods of contraception during treatment with Terrosa. If pregnancy occurs, Terrosa should be discontinued (see Section 4.6 Fertility, Pregnancy and Lactation).
Serum uric acid.
Teriparatide may cause small increases in serum uric acid concentrations. In clinical trials, 2.8% of teriparatide patients had an elevated serum uric acid concentrations compared to 0.7% of placebo patients. However, the hyperuricaemia did not result in an increase in gout, urolithiasis or arthralgia.
Anti-PTH antibody formation.
Anti-PTH antibodies, while apparently clinically irrelevant and only occurring in a small number of treated individuals, have the potential to interfere with the serum PTH estimations.
PTH receptors.
As is generally known, PTH/PTH-related peptide receptors are on multiple tissues. There was no increase in non-osseous tumours in the two 24-month (lifetime) rat studies and in the two 18-month primate studies. There was no increase in incidence of any specific cancer or cancer overall in 2,074 patients in long-term clinical studies or in follow-up studies conducted in a number of these patients for a median of 18 months after teriparatide treatment. Osteosarcoma is a very rare cancer that occurs in 4 out of every million people each year. None of the patients in the clinical trials or post-treatment follow-up developed osteosarcomas.
Other.
New or worsened spinal stenosis was observed in 2 (0.3%) patients who received placebo, 3 (0.4%) patients who received teriparatide 20 microgram, and 3 (0.4%) patients who received teriparatide 40 microgram. One patient who received teriparatide 20 microgram had worsening conductive hearing loss. One patient who received teriparatide 40 microgram required removal of a bone spur and another patient receiving teriparatide 40 microgram required surgical removal of a hyperostosis.4.5 Interactions with Other Medicines and Other Forms of Interactions
No clinically relevant drug interactions have been identified in studies administering teriparatide 40 microgram (twice the recommended dose of teriparatide).
Digoxin.
In a study of 15 healthy subjects administered digoxin daily to steady state, a single teriparatide dose did not alter the effect of digoxin on the systolic time interval (from electrocardiographic Q-wave onset to aortic valve closure, a measure of digoxin's calcium-mediated cardiac effect). However, sporadic case reports have suggested that hypercalcaemia may predispose patients to digitalis toxicity. Because teriparatide transiently increases serum calcium, teriparatide should be used with caution in patients taking digoxin.
Hydrochlorothiazide.
In a study of healthy subjects, the co-administration of 25 mg hydrochlorothiazide with teriparatide did not affect the serum calcium response to teriparatide 40 microgram. The 24-hour urine excretion of calcium was reduced by a clinically insignificant amount (15%).
Frusemide.
In a study of healthy subjects and patients with mild, moderate and severe renal insufficiency (creatinine clearance 13 to 72 mL/min), co-administration of intravenous frusemide (20 to 100 mg) with teriparatide 40 microgram resulted in small, clinically insignificant increases in serum calcium (2%) and in 24-hour urine calcium (37%).
Calcium channel antagonists.
In a study of women with hypertension treated with an extended release preparation of either diltiazem, nifedipine or felodipine, the blood pressure observed after injection of teriparatide 40 microgram was similar when administered alone or in combination with the long-acting calcium channel antagonists.
Atenolol.
In a study of women with hypertension treated with atenolol, the blood pressure observed after injection of teriparatide 40 microgram was similar when administered alone or in combination with atenolol.
Raloxifene.
In a study of healthy postmenopausal women, the co-administration of raloxifene with teriparatide 40 microgram did not alter the effects of teriparatide on serum or urine calcium or on clinical adverse events.
Anti-coagulants.
While this has not been studied, co-administration of anti-coagulants would not be expected to alter the effects of teriparatide. Patients co-administering anti-coagulants and teriparatide need to be advised to take appropriate precautions against the formation of haematomas at the injection sites.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Teriparatide had no adverse effects on fertility of male or female rats at doses up to 300 microgram/kg/day S.C. (about 120 times the human dose based on body surface area). In juvenile rats, treatment with teriparatide was associated with degeneration of the testes at doses ≥ 10 microgram/kg/day S.C. (about 4 times the human dose based on body surface area). Teriparatide should not be used in paediatric patients or young adults (see Section 4.4 Special Warnings and Precautions for Use).
(Category B3)
Category B3 - Drugs which have been taken by only a limited number of pregnant women and women of childbearing age, without an increase in the frequency of malformation or other direct or indirect harmful effects on the human fetus having been observed. Studies in animals have shown evidence of an increased occurrence of fetal damage, the significance of which is considered uncertain in humans.
In pregnant rats given subcutaneous teriparatide doses up to 1000 microgram/kg/day, there were no findings. In pregnant mice given subcutaneous doses of ≥ 30 microgram/kg/day (6 times the human dose based on body surface area) from gestation Day 6 through 15, the foetuses showed an increased incidence of skeletal deviations or variations (interrupted rib, extra vertebra or rib).
Developmental effects in a perinatal/postnatal study in pregnant rats given subcutaneous doses of teriparatide from gestation Day 6 through postpartum Day 20 included mild growth retardation in female offspring at doses of 225 microgram/kg/day (approximately 95 times the human dose based on BSA) and in male offspring at 1000 microgram/kg/day (420 times the human dose based on BSA). There was also reduced motor activity in both male and female offspring at 1000 microgram/kg/day. There were no developmental or reproductive effects in rats at a dose of 30 microgram/kg (12 times the human dose based on BSA).
The effects of teriparatide on the human foetus have not been studied. Terrosa should not be used in pregnant women. See Section 4.3 Contraindications.
Contraception and women of childbearing potential.
Women of childbearing potential should use effective methods of contraception during use of Terrosa. If pregnancy occurs Terrosa should be discontinued (see Section 4.3 Contraindications).
It is not known whether teriparatide is excreted in human milk. Terrosa should not be administered to women who are breast-feeding their children (see Section 4.3 Contraindications).4.7 Effects on Ability to Drive and Use Machines
Teriparatide has no or negligible influence on the ability to drive and use machines. Transient, orthostatic hypotension or dizziness was observed in some patients. These patients should refrain from driving or the use of machines until symptoms have subsided.
4.8 Adverse Effects (Undesirable Effects)
Adverse effects observed with Forteo.
The safety of teriparatide has been evaluated in 21 clinical trials in over 2,800 women and men. Four long-term, Phase 3 clinical trials included one large placebo-controlled, double-blind multinational trial with 1,637 postmenopausal women, one placebo-controlled, double-blind multinational trial with 437 men and two active-controlled trials including 393 postmenopausal women. Teriparatide doses ranged from 5 to 100 microgram/day in short-term trials and 20 to 40 microgram/day in the long-term trials. A total of 1,970 of the patients studied received teriparatide, including 738 patients at 20 microgram/day and 1,107 patients at 40 microgram/day. In the long-term clinical trials, 1,137 patients were exposed to teriparatide for greater than 1 year (500 at 20 microgram/day and 637 at 40 microgram/day). The maximum exposure duration to teriparatide was 2 years. Adverse events associated with Forteo were usually mild and generally did not require discontinuation of therapy.
In the two Phase 3, placebo-controlled clinical trials in men and postmenopausal women, early discontinuation due to an adverse event occurred in 5.6% of patients on placebo and 7.1% of patients on Forteo. Adverse events considered to be related to Forteo therapy were nausea and leg cramps.
Table 1 lists adverse events occurring in the Phase III, placebo-controlled clinical trials in postmenopausal women and in men at a frequency ≥ 2.0% in the Forteo groups and in more Forteo-treated patients than in placebo-treated patients. Adverse events are shown without attributing causality.

Immunogenicity.
In a large clinical trial, antibodies that cross-reacted with teriparatide were detected in 2.8% of female patients receiving Forteo. Generally, antibodies were first detected following 12 months of treatment and diminished after withdrawal of therapy. There were no hypersensitivity reactions, allergic reactions, effects on serum calcium, or effects on bone mineral density (BMD) response, which indicates that the antibodies did not cause any clinically significant adverse effects.
Spontaneous data.
The following table (Table 2) of adverse reactions is based on post-marketing spontaneous reports since market introduction. The following convention has been used for the classification of the adverse reactions: very common (> 1/10), common (> 1/100, < 1/10), uncommon (> 1/1000, < 1/100), rare (> 1/10,000, < 1/1000), very rare (< 1/10,000).
 There has been a report of metastatic osteosarcoma with subsequent fatal outcome in a 72 year old woman with osteoporosis and low back pain who had received teriparatide for 14 months prior to presentation. Causality cannot be established on the basis of this single case and a surveillance program continues. Osteosarcoma occurs at a rate of approximately 4 in one million per year (1 in 250,000 per year) in the general population over 60 years old and at the same rate in women over the age of 70 years. At present it is not known if humans treated with teriparatide have an increased risk of osteosarcoma. However, post-marketing data in humans has not identified an increased risk.
There has been a report of metastatic osteosarcoma with subsequent fatal outcome in a 72 year old woman with osteoporosis and low back pain who had received teriparatide for 14 months prior to presentation. Causality cannot be established on the basis of this single case and a surveillance program continues. Osteosarcoma occurs at a rate of approximately 4 in one million per year (1 in 250,000 per year) in the general population over 60 years old and at the same rate in women over the age of 70 years. At present it is not known if humans treated with teriparatide have an increased risk of osteosarcoma. However, post-marketing data in humans has not identified an increased risk.
Comparability of Terrosa with Forteo.
In the clinical trials of Terrosa 54 healthy premenopausal females and 125 patients with osteoporosis at high risk of fracture, a total of 179 subjects were exposed to Terrosa and the safety profile for Terrosa was consistent with that observed for Forteo.
Immunogenicity.
Comparative immunogenicity data was generated in the Phase III comparative clinical study (Study RGB1023O31) of Terrosa and Forteo. Anti-teriparatide antibody levels were investigated at baseline and Week 52 or at the time of discontinuation to provide comparative data on the immunogenic potential of Terrosa. No subjects in the Terrosa group and 2 subjects (1.6%) in the Forteo group were positive for anti-teriparatide antibody. The immunogenic potential of Terrosa did not exceed that of the reference product Forteo. For study details please see Section 5.1 Pharmacodynamic Properties.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Signs and symptoms.
No cases of overdose were reported during clinical trials. Teriparatide has been safely administered in single doses of up to 100 microgram. In a clinical study, doses of 60 microgram/day for 6 weeks were safely tolerated. The effects of overdose that might be expected include delayed hypercalcaemia and risk of orthostatic hypotension. Nausea, vomiting, dizziness, and headache might also occur.
Overdose experience based on post-marketing spontaneous reports.
In post-marketing spontaneous reports, there have been cases of medication error in which the entire contents (up to 800 microgram) of the teriparatide cartridge have been administered as a single dose. Transient events reported have included nausea, weakness/lethargy and hypotension. In some cases, no adverse events occurred as a result of the overdose. No fatalities associated with overdose have been reported.
In single-dose rodent studies using subcutaneous injection of teriparatide, no mortality was seen in rats given doses of 1000 microgram/kg (526 times the human dose based on body surface area) or in mice given 10,000 microgram/kg (2,635 times the human dose).
Overdose management.
There is no specific antidote for teriparatide. Treatment of suspected overdose should include discontinuation of teriparatide, monitoring of serum calcium, and implementation of appropriate supportive measures, such as hydration.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Osteoporosis is characterised by low bone mass and microarchitectural deterioration of bone tissue, leading to bone fragility and an increase in the risk of vertebral and non-vertebral fractures. The diagnosis of osteoporosis may be confirmed by the finding of low bone mass or the presence or history of osteoporotic fracture. While non-vertebral fractures are usually clinically apparent, vertebral fractures also may be manifested by back pain, height loss or kyphosis.
Mechanism of action.
Endogenous 84-amino-acid parathyroid hormone (PTH) is the primary regulator of calcium and phosphate metabolism in bone and kidney. Teriparatide is the active fragment (1-34) of endogenous human parathyroid hormone, manufactured using recombinant DNA technology.
Physiological actions of PTH include regulation of bone metabolism, renal tubular reabsorption of calcium and phosphate, and intestinal calcium absorption. The biological actions of PTH and teriparatide are mediated through binding to specific PTH cell surface receptors. Teriparatide binds to these receptors with similar affinity as PTH and has the same actions in bone and kidney as PTH. Like endogenous PTH, teriparatide is not expected to accumulate in bone or other tissues.
Pharmacodynamic effects.
The skeletal effects of teriparatide depend upon the pattern of systemic exposure. Once daily administration of teriparatide increases apposition of new bone on trabecular and cortical (endosteal and periosteal) bone surfaces by preferential stimulation of osteoblastic activity over osteoclastic activity. In contrast, continuous excess of endogenous PTH, as occurs in hyperparathyroidism, may be detrimental to the skeleton because bone resorption may be stimulated more than bone formation.
In monkey studies, teriparatide improved trabecular microarchitecture and increased bone mass and strength by stimulating new bone formation in both cancellous and cortical bone. In humans, teriparatide affects calcium and phosphorus metabolism in a pattern consistent with the known actions of endogenous PTH.
Clinical trials.
Clinical trials with Forteo.
The clinical program included treatment studies in women and men with osteoporosis. Postmenopausal women were treated for up to 24 months to evaluate effects on vertebral fractures. Men were treated for up to 14 months to evaluate the effect on BMD. Of the women and men who participated in the Forteo treatment studies, 1,930 have been systematically observed for 18 months in a post-treatment follow-up study.
Treatment of postmenopausal women with osteoporosis. The pivotal study included 1637 postmenopausal women (mean age 69.5 years). At baseline, ninety percent of the patients had one or more vertebral fractures. All patients received 1000 mg of calcium per day and at least 400 IU of vitamin D per day. Results from a treatment period of up to 24 months (median 19 months), with teriparatide, demonstrate significant anti-fracture efficacy.
Effect on vertebral fractures.
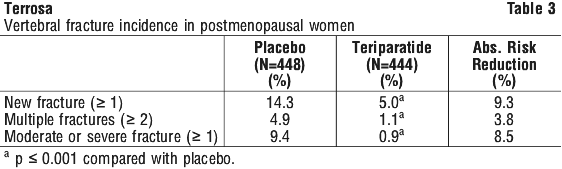
Teriparatide, relative to placebo, given for a median of 19 months, significantly reduced the risk and severity of new vertebral fractures in postmenopausal women with osteoporosis. The relative risk for the incidence of 1 or more new vertebral fractures was reduced by 65% and multiple fractures by 77% with teriparatide treatment (Table 3 includes data on absolute risk reduction). Eleven women would need to be treated with teriparatide for a median of 19 months to prevent one or more new vertebral fractures.
Effect on non-vertebral fractures.
Teriparatide significantly reduced (by 53%) the overall incidence of non-vertebral fragility fractures including wrist, ribs, ankle, humerus, hip, foot, pelvis and others (see Figure 1).
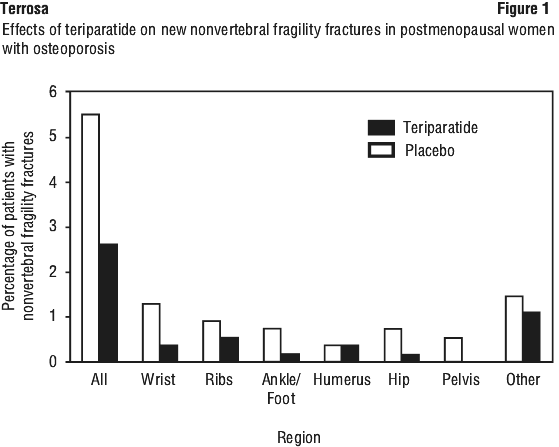
Effect on BMD.
Teriparatide rapidly increased lumbar spine BMD. Significant increases were seen as early as 3 months and continued throughout the treatment period, as shown in Figure 2. After a median treatment period of 19 months, BMD had increased 9% and 4% in the lumbar spine and total hip, respectively, compared with placebo (p < 0.001). Teriparatide was effective regardless of age, baseline rate of bone turnover and baseline BMD.

Effect on back pain.
Teriparatide significantly reduced the incidence and severity of back pain. In women with postmenopausal osteoporosis, there was a significant (p = 0.017) 26% reduction in the spontaneous reports of new or worsened back pain compared to placebo.
Effects on height loss.
For the 86 postmenopausal women who experienced vertebral fractures, those treated with teriparatide had significantly less height loss when compared to placebo (p = 0.001).
Bone histology.
The effects of teriparatide on bone histology were evaluated in iliac crest biopsies of 61 postmenopausal women treated for up to 24 months with placebo or teriparatide 20 microgram or 40 microgram per day. The increases in BMD and resistance to fracture achieved with teriparatide occurred without evidence of cellular toxicity or adverse effects on bone architecture or mineralisation. The findings in human bone samples paralleled those seen in preclinical primate studies.
Post-treatment fracture efficacy.
Following treatment with teriparatide, 1,262 postmenopausal women from the pivotal trial enrolled in a post-treatment follow-up study. After 18 months, approximately 50% of the women in each former treatment group had begun an approved osteoporosis therapy (not including teriparatide) at the discretion of their physician. All women were offered 1000 mg of calcium per day and at least 400 IU of vitamin D per day.
During a median of 18 months following discontinuation of teriparatide treatment, there was a significant 40% reduction in relative risk for new vertebral fractures in women previously treated with teriparatide, compared to placebo. The relative risk reduction was similar for women with and without osteoporosis treatment, 41% and 37%, respectively. During the same observation period, there was a 42% risk reduction for nonvertebral fragility fractures in women previously treated with teriparatide, compared with placebo.
Data from this study demonstrate that regardless of the follow-up treatment options, fracture risk was reduced for women previously treated with teriparatide.
A 24-month, randomised, double-blind, comparator-controlled Phase 4 study included 1,360 postmenopausal women with established osteoporosis. 680 subjects were randomised to Forteo and 680 subjects were randomised to oral risedronate 35 mg/week. At baseline, the women had a mean age of 72.1 years and a median of 2 prevalent vertebral fractures; 57.9% of patients had received previous bisphosphonate therapy and 18.8% took concomitant glucocorticoids during the study. 1,013 (74.5%) patients completed the 24-month follow-up. The mean (median) cumulative dose of glucocorticoid was 474.3 (66.2) mg in the teriparatide arm and 898.0 (100.0) mg in the risedronate arm. The mean (median) vitamin D intake for the teriparatide arm was 1408.4 IU/day (1380.4 IU/day) and for the risedronate arm was 1206.4 IU/day (900 IU/day). For those subjects who had baseline and follow-up spine radiographs, the incidence of new vertebral fractures was 28/516 (5.4%) in Forteo and 64/533 (12.0%) in risedronate-treated patients, relative risk (95% CI) = 0.44 (0.29-0.68), p < 0.0001. The cumulative incidence of pooled clinical fractures (clinical vertebral and non-vertebral fractures) was 4.8% in Forteo and 9.8% in risedronate-treated patients, hazard ratio (95% CI) = 0.48 (0.32-0.74), P = 0.0009.
In an open-label study, 503 postmenopausal women with severe osteoporosis and a fragility fracture within the previous 3 years (83% had received previous osteoporosis therapy) were treated with Forteo for up to 24 months. At 24 months, the mean increase from baseline in lumbar spine, total hip and femoral neck BMD was 10.5%, 2.6% and 3.9%, respectively. The mean increase in BMD from 18 to 24 months was 1.4%, 1.2% and 1.6% at the lumbar spine, total hip and femoral neck, respectively.
Male osteoporosis. The efficacy of teriparatide was demonstrated in a double-blind, placebo-controlled clinical study in 437 men with either hypogonadal or idiopathic osteoporosis. All patients received 1000 mg of calcium per day and at least 400 IU of vitamin D per day and were treated for up to 14 months.
In this study, teriparatide rapidly increased lumbar spine BMD in men, with significant increases as early as 3 months. This increase continued throughout the treatment period, as shown in Figure 3. After a median treatment period of 11 months, BMD in the spine had (on average) increased by 5% and in the hip by 1%, compared to placebo. Increases in BMD were similar in men with hypogonadal or idiopathic osteoporosis. Teriparatide was effective regardless of age, baseline rate of bone turnover and baseline BMD.
All male patients presenting with osteoporosis should be checked for primary or secondary hypogonadism, investigated and treated appropriately as a prerequisite.
 Glucocorticoid-induced osteoporosis. The efficacy of Forteo in men and women (N = 428) receiving sustained systemic glucocorticoid therapy (equivalent to 5 mg or greater of prednisone for at least 3 months) was demonstrated in a 36 month (18-month primary phase plus 18-month continuation phase), randomised, double-blind, comparator-controlled study (alendronate 10 mg/day). Twenty-eight percent of patients had one or more radiographic vertebral fractures at baseline. All patients were offered 1000 mg calcium per day and 800 IU vitamin D per day.
Glucocorticoid-induced osteoporosis. The efficacy of Forteo in men and women (N = 428) receiving sustained systemic glucocorticoid therapy (equivalent to 5 mg or greater of prednisone for at least 3 months) was demonstrated in a 36 month (18-month primary phase plus 18-month continuation phase), randomised, double-blind, comparator-controlled study (alendronate 10 mg/day). Twenty-eight percent of patients had one or more radiographic vertebral fractures at baseline. All patients were offered 1000 mg calcium per day and 800 IU vitamin D per day.
This study included postmenopausal women (N = 277), premenopausal women (N = 67), and men (N = 83). At baseline, the postmenopausal women had a mean age of 61 years, mean lumbar spine BMD T score (number of standard deviations above or below the mean in healthy young women) of -2.7, median prednisone equivalent dose of 7.5 mg/day, and 34% had one or more radiographic vertebral fractures; premenopausal women had a mean age of 37 years, mean lumbar spine BMD T score of -2.5, median prednisone equivalent dose of 10 mg/day, and 9% had one or more radiographic vertebral fractures; and men had a mean age of 57 years, mean lumbar spine BMD T score of -2.2, median prednisone equivalent dose of 10 mg/day, and 24% had one or more radiographic vertebral fractures.
Effects on vertebral and non-vertebral BMD.
The primary objective was the change in lumbar spine BMD from baseline to the 18-month endpoint (last observation carried forward) in men and women combined. Sixty-nine percent of patients completed the 18-month primary phase. At the 18-month endpoint (men and women combined), Forteo increased lumbar spine BMD (7.2%) significantly more than alendronate (3.4%) (p < 0.001).
Figure 4 shows the time course of mean percent change from baseline in lumbar spine BMD through 36 months for men and women combined. There was a significant difference between groups at all measured timepoints and endpoint. At 36 months (Figure 4) the mean percent change from baseline in lumbar spine BMD was 11.0% in the Forteo group versus 5.3% in the alendronate group, a difference of 5.7% (p < 0.001).
 Table 4 presents the mean percent change in lumber spine BMD in the women only subgroup.
Table 4 presents the mean percent change in lumber spine BMD in the women only subgroup.
 In men and women combined, changes from baseline in femoral neck BMD were significantly greater in the Forteo compared with the alendronate group at all timepoints and at endpoint (Figure 5). The mean percent change in femoral neck BMD from baseline to endpoint was 5.1% in the Forteo group compared with 2.6% in the alendronate group, (p < 0.001).
In men and women combined, changes from baseline in femoral neck BMD were significantly greater in the Forteo compared with the alendronate group at all timepoints and at endpoint (Figure 5). The mean percent change in femoral neck BMD from baseline to endpoint was 5.1% in the Forteo group compared with 2.6% in the alendronate group, (p < 0.001).
 Forteo group compared with the alendronate group at all timepoints and at endpoint (Figure 6). The mean increase in total hip BMD from baseline to endpoint was 4.4% in the Forteo group versus 2.2% in the alendronate group (p < 0.001).
Forteo group compared with the alendronate group at all timepoints and at endpoint (Figure 6). The mean increase in total hip BMD from baseline to endpoint was 4.4% in the Forteo group versus 2.2% in the alendronate group (p < 0.001).
 In premenopausal women, the increase in BMD from baseline to endpoint at 36 months was significantly greater in the Forteo group compared with the alendronate group at the lumbar spine (4.6% versus -0.9%; p = 0.017) and total hip (4.8% versus 1.5%; p = 0.026). However, no significant effect on fracture rates was demonstrated in premenopausal women.
In premenopausal women, the increase in BMD from baseline to endpoint at 36 months was significantly greater in the Forteo group compared with the alendronate group at the lumbar spine (4.6% versus -0.9%; p = 0.017) and total hip (4.8% versus 1.5%; p = 0.026). However, no significant effect on fracture rates was demonstrated in premenopausal women.
Analysis of vertebral and non-vertebral fractures.
At 18 months, analysis of spinal X-rays from 165 alendronate patients and 171 Forteo patients showed that 10 patients in the alendronate group (6.1%) had experienced a new vertebral fracture compared with 1 patient in the Forteo group (0.6%). In addition, 9 patients in the alendronate group (4.2%) had experienced a nonvertebral fracture compared with 12 patients in the Forteo group (5.6%).
Table 5 summarises the incident fractures at 36 months in men and women combined.

Effects on markers of bone turnover.
In patients with glucocorticoid-induced osteoporosis, daily administration of Forteo stimulated new bone formation as shown by increases from baseline in the serum concentration of biochemical markers of bone formation including bone-specific alkaline phosphatase (BSAP), procollagen I carboxy-terminal propeptide (PICP), and amino-terminal propeptide of type I collagen (P1NP) (see Table 6). Forteo also stimulated bone resorption as shown by increases from baseline in serum concentrations of C-terminal telopeptide of type I collagen (CTX). Alendronate 10 mg/day induced decreases from baseline in the serum concentration of BSAP, PICP, P1NP and CTX (see Table 6). The effects of Forteo on bone turnover markers in patients with glucocorticoid-induced osteoporosis were qualitatively similar to the effects in postmenopausal women with osteoporosis not taking glucocorticoids.

Comparability of Terrosa with Forteo.
Study RGB1023O31.
Therapeutic equivalence of Terrosa and Forteo was demonstrated in a Japanese, multicentre, randomised, non-inferiority, active drug controlled, rater-blinded, parallel-group comparative phase III study in men and post-menopausal women with osteoporosis at high risk of fracture (a total of 250 subjects). Subjects were administered Terrosa or Forteo (teriparatide 20 microgram/80 microL solution for injection) daily via S.C. injection for 52 weeks.
The percent change (mean ± standard deviation [SD]) in lumbar spine (L2-L4) BMD at Week 52, which served as the primary endpoint of the study, was 8.94% ± 6.19% in the Terrosa group and 9.65% ± 6.22% in the Forteo group. The (adjusted) difference in the mean between the Terrosa and Forteo groups was -0.65% (2-sided 95% CI: -2.17% to 0.87%). The 2-sided 95% CI was within the range of the pre-specified equivalence margin (± 2.8).
There were no substantial differences between the Terrosa and the Forteo groups in terms of the secondary endpoints of the absolute change in lumbar spine (L2-L4) BMD, the percent and absolute changes in lumbar spine (L1-L4) BMD, femoral neck BMD and proximal femoral BMD, the percent and absolute changes in bone metabolic marker of P1NP and the incidence of vertebral or non-vertebral fractures.
5.2 Pharmacokinetic Properties
Absorption.
After subcutaneous (S.C.) injection, teriparatide has an absolute bioavailability of 95% (95% CI 0.824 - 1.07). Absorption and elimination are rapid. The half-life of teriparatide in serum is 5 minutes when administered by intravenous injection and approximately 1 hour when administered by subcutaneous injection. The longer half-life following subcutaneous administration reflects the time required for absorption from the injection site.
Following a subcutaneous injection of a 20 microgram dose, peak molar concentrations of teriparatide briefly exceed the upper limit of normal for endogenous PTH [65 picogram/mL (7.0 picoM)] by 4- to 5-fold for about 30 minutes and then decline to non-quantifiable concentrations within 3 hours. The mean systemic exposure (endogenous PTH and teriparatide) over 24 hours does not exceed the upper limit of normal and is below the levels found in patients with mild hyperparathyroidism.
Distribution.
Volume of distribution is approximately 1.7 L/kg. Between-subject variability in systemic clearance and volume of distribution is 25% to 50%.
Metabolism.
No metabolism studies have been performed with teriparatide. However, the mechanisms of metabolism of PTH(1-34) and intact PTH have been extensively described. Metabolism of parathyroid hormone is believed to occur predominantly in liver and kidney.
Excretion.
Teriparatide is eliminated through hepatic and extra-hepatic clearance (approximately 62 L/hr in women and 94 L/hr in men). No excretion studies have been performed with teriparatide. However, the mechanism of elimination of PTH(1-34) and intact PTH have been extensively described.
Patient characteristics.
Geriatrics.
No differences in teriparatide pharmacokinetics were detected with regard to age (range 31 to 85 years). Dosage adjustment based on age is not required.
Gender.
Systemic exposure to teriparatide is approximately 20% to 30% lower in men than in women. There were, however, no gender differences with respect to safety, tolerability or pharmacodynamic responses. Dosage adjustment based on gender is not required.
Renal impairment.
No clinically relevant pharmacokinetic or safety differences were identified in patients with mild, moderate or severe chronic renal impairment administered a single dose of teriparatide. Dosage adjustment, based on renal function, is not required.
However, patients with renal impairment had reduced calcaemic and calciuric responses to teriparatide. Long-term safety and efficacy have not been evaluated in patients with serum creatinine concentrations > 177 micromol/L.
Heart failure.
No clinically relevant pharmacokinetic, blood pressure, pulse rate or other safety differences were identified in patients with stable heart failure (New York Heart Association Class I to III and additional evidence of cardiac dysfunction) administered two 20 microgram doses of teriparatide. Dosage adjustment based on the presence of mild or moderate heart failure is not required. There are no data from patients with severe heart failure.
Hepatic impairment.
Safety and efficacy have not been evaluated in patients with hepatic impairment. Preclinical data indicate that hepatic Kupffer cells are the primary site of metabolism for teriparatide. It is unlikely that disease states in which hepatocyte function is impaired will have a clinically significant effect on systemic exposure to teriparatide).
Comparative bioavailability (bioequivalence).
The pharmacokinetic equivalence of the biosimilar Terrosa with the reference Forteo (EU trade name for teriparatide) was demonstrated in a double-blind, randomised, 2-way crossover Phase I study RGB10-001 in 54 healthy female volunteers. The pharmacokinetic parameters are summarised in Table 7.

5.3 Preclinical Safety Data
Genotoxicity.
Teriparatide was not genotoxic in assays for gene mutations (Ames test and mouse lymphoma assay in vitro) and chromosomal damage (Chinese hamster ovary cells in vitro and the mouse micronucleus test in vivo).
Carcinogenicity.
Two carcinogenicity bioassays were conducted in rats. In the first study, male and female rats were given daily subcutaneous teriparatide injections of 5, 30, or 75 microgram/kg/day for 24 months from 2 months of age. These doses resulted in systemic exposures that were, respectively, 3, 20, and 60 times higher than the systemic exposure observed in humans following a subcutaneous dose of 20 microgram (based on AUC comparison). Teriparatide treatment resulted in a marked dose-related increase in the incidence of osteosarcoma, a rare malignant bone tumour, in both male and female rats. Osteosarcomas were observed at all doses, occurred after 17 to 20 months of treatment, and reached an incidence of 38% to 52% in the high-dose groups.
Teriparatide also caused increased incidences of osteoblastoma and osteoma in both sexes. No osteosarcomas, osteoblastomas or osteomas were observed in untreated control rats. The bone tumours in rats occurred in association with a large increase in bone mass and focal osteoblast hyperplasia.
The second 2-year study was carried out in order to determine the effect of treatment duration and animal age on the development of bone tumours. Female rats were treated for different periods between 2 and 26 months of age with subcutaneous doses of 5 and 30 microgram/kg (equivalent to 3 and 20 times the human exposure at the 20 microgram dose, based on AUC). The study showed that the occurrence of osteosarcoma, osteoblastoma and osteoma was dependent upon dose and duration of exposure. Bone tumours were observed when immature 2-month old rats were treated with 30 microgram/kg/day for 6 or 24 months. Bone tumours were also observed when mature 6-month old rats were treated with 30 microgram/kg/day for 6 or 20 months. Tumours were not detected when mature 6-month old rats were treated with 5 microgram/kg/day for 6 or 20 months. The results did not demonstrate a difference in susceptibility to bone tumour formation, associated with teriparatide treatment, between mature and immature rats. The relevance of these rat findings to humans is uncertain.
No bone neoplasms or preneoplastic lesions were found in monkeys treated with teriparatide S.C. for 18 months, and then observed for a further 3 years, at a dose yielding 5-fold clinical exposure levels (based on AUC data).6 Pharmaceutical Particulars
6.1 List of Excipients
Glacial acetic acid, mannitol, metacresol, sodium acetate trihydrate, hydrochloric acid (for pH adjustment), sodium hydroxide (for pH adjustment), water for injections.
This medicinal product contains less than 1 mmol sodium (23 mg) per dosage unit, that is to say essentially "sodium-free".
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
To avoid medication errors, make sure that the date when starting to use a new pre-filled pen is at least 28 days before its expiry.
Chemical in-use stability has been demonstrated for 28 days at 2°C to 8°C.
6.4 Special Precautions for Storage
Store at 2°C to 8°C. Refrigerate. Do not freeze.
Do not use Terrosa if it has been frozen.
The dose can be used immediately following removal from the refrigerator.
The pre-filled pen should be returned to the refrigerator immediately after use. Keep the pen cap on the prefilled pen in order to protect from light.
Do not store the pre-filled pen with the needle attached.
From a microbiological point of view, once opened, the product may be stored for a maximum of 28 days within its shelf life at 2°C to 8°C.
Terrosa should not be used if the solution is cloudy, coloured or contains visible particle.
6.5 Nature and Contents of Container
Each pre-filled pen contains a 3 mL cartridge (siliconised Type I glass), with a plunger stopper and disc seal (aluminium and rubber liner seals), packed in a carton.
One cartridge of 2.4 mL of solution for injection contains 600 microgram of teriparatide (corresponding to 250 microgram/mL).
Each pre-filled pen contains 2.4 mL of solution corresponding to 28 doses of 20 microgram (per 80 microlitres).
Pack sizes.
Terrosa is available in packs of 1 or 3 pre-filled pens.
Not all pack sizes may be marketed.
6.6 Special Precautions for Disposal
Terrosa pre-filled pen is for multidose use. After 28 days of treatment the pre-filled pen should be properly disposed of after 28 days of first use, even if it is not completely empty.
In Australia, any unused medicine or waste should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Pharmacotherapeutic group: Calcium homeostasis, parathyroid hormones and analogues.
ATC code: H05AA02.
Teriparatide has a molecular weight of 4115.1309 Dalton. In other sources, the mass for teriparatide is given with 4118 Dalton.
Chemical structure.
Teriparatide, rhPTH(1-34), produced in E. coli, using recombinant DNA technology, is identical to the 34-N-terminal amino acid sequence of endogenous human parathyroid hormone.
 Terrosa is manufactured using recombinant DNA technology.
Terrosa is manufactured using recombinant DNA technology.
CAS number.
The Chemical Abstract Service (CAS) Registry Number for teriparatide is 52232-67-4.7 Medicine Schedule (Poisons Standard)
Schedule 4 Prescription only medicine.
Summary Table of Changes
