
SUMMARY CMI
TEVETEN®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using TEVETEN?
TEVETEN contains the active ingredient eprosartan. TEVETEN is used to lower blood pressure which doctors call hypertension.
For more information, see Section 1. Why am I using TEVETEN? in the full CMI.
2. What should I know before I use TEVETEN?
Do not use if you have ever had an allergic reaction to eprosartan or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use TEVETEN? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with TEVETEN and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use TEVETEN?
- Your doctor will decide what dose you should receive.
- The usual starting dose is one 600 mg tablet per day. However, some people may need a lower starting dose.
More instructions can be found in Section 4. How do I use TEVETEN? in the full CMI.
5. What should I know while using TEVETEN?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using TEVETEN? in the full CMI.
6. Are there any side effects?
Tell your doctor if you notice any of the following and they worry you: feeling light-headed, dizzy or faint, headache, cough, 'flu-like' symptoms, feeling sick (nausea), or vomiting, stomach pain or discomfort, unusual tiredness or weakness, fatigue, joint pain, urinary tract infection or fast or slow heartbeats.
Tell your doctor immediately if you notice any of the following: hives or itching, reddening of the skin or other unusual skin reactions that worry you.
Go to Accident and Emergency at your nearest hospital, if you notice any of the following: swelling of limbs, face, eyes, inside the nose, mouth or throat, shortness of breath, breathing or swallowing difficulties, unusual tiredness or weakness that is sudden and severe or chest pain.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
TEVETEN®
Active ingredient(s): Eprosartan mesilate
Consumer Medicine Information (CMI)
This leaflet provides important information about using TEVETEN. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using TEVETEN.
Where to find information in this leaflet:
1. Why am I using TEVETEN?
2. What should I know before I use TEVETEN?
3. What if I am taking other medicines?
4. How do I use TEVETEN?
5. What should I know while using TEVETEN?
6. Are there any side effects?
7. Product details
1. Why am I using TEVETEN?
TEVETEN contains the active ingredient eprosartan.
TEVETEN belongs to a group of medicines called angiotensin-II receptor antagonists. It works by blocking the action of a substance called angiotensin II. As a result, TEVETEN helps relax the blood vessels and helps lower your blood pressure.
TEVETEN is used to lower high blood pressure, which doctors call hypertension.
There are usually no symptoms of hypertension. The only way of knowing if you have hypertension is to have your blood pressure checked regularly. If high blood pressure is not treated it can lead to serious health problems, including stroke, heart disease and kidney failure.
Your doctor may have prescribed TEVETEN for another use.
Ask your doctor if you have any questions about why TEVETEN has been prescribed for you.
There is no evidence that TEVETEN is addictive.
There is no specific information available to recommend the use of TEVETEN in children.
2. What should I know before I use TEVETEN?
Warnings
Do not use TEVETEN if:
- you are allergic to eprosartan, or any of the ingredients listed at the end of this leaflet.
- Signs of allergic reaction may include itchy skin rash, shortness of breath and swelling of the face or tongue.
Always check the ingredients to make sure you can use this medicine.
- Tell your doctor or pharmacist if you have taken TEVETEN before and became unwell.
- Do not take TEVETEN if you have serious problems with your kidneys.
- Do not take TEVETEN if you have diabetes or kidney problems and you are taking aliskiren (a medicine used to treat high blood pressure).
- Do not take TEVETEN if the expiry date (EXP) printed on the pack has passed.
- If you take it after the expiry date has passed, it may not work as well. - Do not take TEVETEN if the packaging is torn or shows signs of tampering or if the tablets show any signs of visual deterioration.
Check with your doctor if you:
- have had an allergic reaction to any medicine that you have taken previously to treat your high blood pressure.
- have allergies to any other medicines or any other substances, such as dyes, foods or preservatives.
- have any of these medical conditions:
- liver problems
- kidney problems
- heart problems
- adrenal problems
- elevated potassium levels - have experienced recent excessive vomiting or diarrhoea.
- are on a salt restricted diet.
- have hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Do not take TEVETEN if you are pregnant, or think you may be pregnant.
Check with your doctor if you are pregnant or intend to become pregnant.
Do not take TEVETEN if you are breastfeeding, or wish to breastfeed.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and TEVETEN may interfere with each other. These include but are not limited to:
- other medicines to lower your blood pressure, especially ACE inhibitors or medicines that contain aliskiren.
- diuretics (fluid or water tablets).
- non-steroidal anti-inflammatory drugs (NSAIDs), medicines used to relive pain, swelling and other symptoms of inflammation.
- lithium, a medicine used to treat mood swings and some types of depression.
- potassium supplements or potassium-containing salt substitutes.
These medicines may be affected by TEVETEN, or may affect how well it works. Other medicines used to treat high blood pressure may have an additive effect with TEVETEN in lowering your blood pressure. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor or pharmacist will be able to tell you more about which medicines can be taken with TEVETEN.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect TEVETEN.
4. How do I use TEVETEN?
How much to take
Follow the directions your doctor has given you on how many tablets to take each day.
These will be printed on the pharmacy label on the box.
- The usual starting dose of TEVETEN is one 600 mg tablet per day. However, some people may need a lower starting dose. Your doctor may recommend a lower starting dose if you have liver or kidney problems, or if you are over 60 years of age, or you are dehydrated or on diuretics (fluid tablets).
- TEVETEN can take 2 to 3 weeks to work fully.
- Follow the instructions provided and use TEVETEN until your doctor tells you to stop.
When to take TEVETEN
- Take TEVETEN regularly at the same time each day.
- Taking your medicine at the same time each day will give the best effect. It will also help you to remember when to take your medicine.
How to take TEVETEN
- Swallow TEVETEN tablets with a full glass of water or another liquid.
- TEVETEN tablets can be taken with or without food. Do not chew the tablets.
How long to take it
- Continue taking TEVETEN for as long as your doctor prescribes it.
- TEVETEN helps control your high blood pressure, but does not cure it. Therefore, TEVETEN must be taken every day.
If you forget to use TEVETEN
TEVETEN should be used regularly at the same time each day.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to. Otherwise, take it as soon as you remember, and then go back to taking your tablets as you would normally.
DO NOT take two doses within 6 hours of each other.
Do not take a double dose to make up for the dose you missed.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you use too much TEVETEN
If you think that you have used too much TEVETEN, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(Australia telephone 13 11 26) for advice, or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
If possible, show the doctor the pack of tablets.
5. What should I know while using TEVETEN?
Things you should do
- Be sure to keep all your appointments with your doctor so that your progress can be checked.
- If you become pregnant while taking TEVETEN, tell your doctor immediately.
- If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly.
- Make sure you drink enough water during exercise and hot weather when you are taking TEVETEN, especially if you sweat a lot.
- If you feel faint or light-headed during exercise or hot weather, speak to your doctor. - Tell your doctor if you have excessive vomiting and/or diarrhoea while taking TEVETEN.
- If you start or stop any new medicine, including any that you buy without a prescription from your pharmacy, supermarket or health food shop, tell your doctor and pharmacist that you are taking TEVETEN.
- If you plan to have surgery (even at the dentist) that requires a general anaesthetic, tell your doctor or dentist that you are taking TEVETEN.
- Remind any doctor, dentist or pharmacist you visit that you are using TEVETEN.
Things you should not do
- Do not stop taking TEVETEN without checking with your doctor.
- Do not use TEVETEN to treat any other complaints, unless your doctor tells you to.
- Do not give TEVETEN to anyone else even if they have the same condition as you.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how TEVETEN affects you.
As with other medicines used to treat high blood pressure, TEVETEN may cause dizziness or light-headedness in some people, especially after the first dose or when your dose is changed.
Make sure you know how you react to TEVETEN before you drive a car, operate machinery or do anything else that could be dangerous if you are dizzy.
Looking after your medicine
Follow the instructions in the carton on how to take care of your medicine properly.
Keep your tablets in the original pack until it is time to take them.
Keep the pack in a cool, dry place (below 25°C). Do not store TEVETEN in the bathroom, near a sink or leave it in the car on hot days.
Heat and dampness can destroy some medicines.
Keep TEVETEN where young children cannot reach it.
A locked cupboard at least one-and-a half metres above the ground is a good place to store medicines.
Getting rid of any unwanted medicine
If your doctor tells you to stop taking this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
Check with your doctor as soon as possible if you have any problems while taking TEVETEN, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
This medicine helps most people with high blood pressure; however, like other medicines it may cause some side effects.
Do not be alarmed by this list of side effects.
You may not experience any of them. If they occur, most are likely to be minor and temporary. However, some may be serious and need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
These are serious side effects and may be symptoms of an allergic reaction. Allergy to TEVETEN is rare.
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
All possible side effects of TEVETEN may not be currently known.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What TEVETEN 400mg tablet contains
| Active ingredient (main ingredient) | eprosartan mesilate |
| Other ingredients (inactive ingredients) | lactose monohydrate |
| Potential allergens | TEVETEN contains lactose. |
What TEVETEN 600mg tablet contains
| Active ingredient (main ingredient) | eprosartan mesilate |
| Other ingredients (inactive ingredients) | lactose monohydrate |
| Potential allergens | TEVETEN contains lactose. |
Do not take this medicine if you are allergic to any of these ingredients.
What TEVETEN looks like
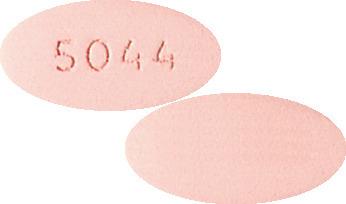
TEVETEN 400 mg tablets are light to moderately pink, oval, film coated tablets marked with “5044” on one side and plain on the other side. (AUST R 64400).
TEVETEN 600 mg tablets are white, capsule-shaped, film-coated tablets marked with “5046” on one side and plain on the other side. (AUST R 73779).
Who distributes TEVETEN
Viatris Pty Ltd
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in January 2022.
TEVETEN® is a Viatris company trade mark.
TEVETEN_cmi\Jan22/00
Published by MIMS March 2022