1 Name of Medicine
Tislelizumab.
2 Qualitative and Quantitative Composition
Each Tevimbra single dose vial contains 100 mg of tislelizumab in 10 mL solution, with a concentration of 10 mg/mL.
Excipient(s) with known effect.
Each mL of concentrate for solution for infusion contains 0.069 mmol (or 1.6 mg) sodium.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Concentrate for solution for infusion.
Clear to slightly opalescent, colorless to slightly yellow solution.
The solution has a pH of approximately 6.5 and an osmolality of approximately 270 to 330 mOsm/kg.
4.1 Therapeutic Indications
Oesophageal squamous cell carcinoma (OSCC).
Tevimbra in combination with platinum-based chemotherapy is indicated for the first-line treatment of patients with unresectable, locally advanced or metastatic oesophageal squamous cell carcinoma with a PD-L1 expression ≥ 1%.
Tevimbra as monotherapy is indicated for the treatment of adult patients with unresectable, recurrent, locally advanced or metastatic oesophageal squamous cell carcinoma after prior chemotherapy.
Non-small cell lung cancer (NSCLC).
Tevimbra in combination with pemetrexed and platinum containing chemotherapy is indicated for the first-line treatment of patients with locally advanced or metastatic non-squamous non-small cell lung cancer (NSCLC), with PD-L1 expression ≥ 50% but no epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) genomic tumour aberrations.
Tevimbra in combination with carboplatin and either paclitaxel or nab-paclitaxel is indicated for the first-line treatment of patients with locally advanced or metastatic squamous NSCLC.
Tevimbra as monotherapy is indicated for the treatment of patients with locally advanced or metastatic NSCLC after prior chemotherapy.
Gastric/gastro-oesophageal junction (G/GOJ) adenocarcinoma.
Tevimbra in combination with platinum and fluoropyrimidine-based chemotherapy, is indicated for the first-line treatment of adult patients with HER-2 negative locally advanced unresectable or metastatic gastric or gastro-oesophageal junction (G/GOJ) adenocarcinoma with a PD-L1 expression ≥ 1%.
Nasopharyngeal carcinoma (NPC).
Tevimbra in combination with gemcitabine and cisplatin is indicated for the first-line treatment of adult patients with recurrent or metastatic nasopharyngeal carcinoma (NPC).4.2 Dose and Method of Administration
Tevimbra should be administered under the supervision of a physician experienced in the use of anti-cancer therapy.
Tevimbra is for single use in one patient only. Discard any residue after use.
Dosage.
The recommended dose of Tevimbra is 200 mg administered once every 3 weeks as an intravenous infusion.
When Tevimbra is used in combination, see Table 1 for the full prescribing information of the combination therapy. When administering Tevimbra in combination with chemotherapy, administer Tevimbra before chemotherapy when both are given on the same day.
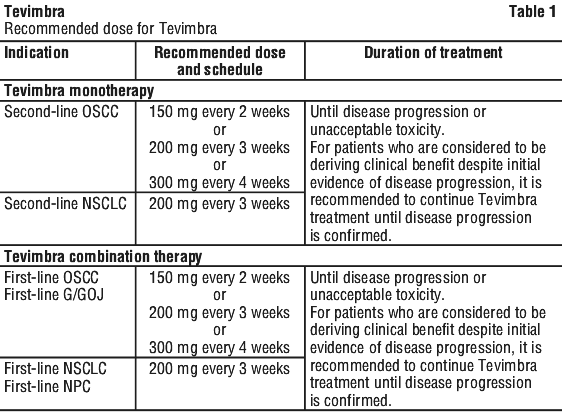
Patient selection.
Prior to the use of tislelizumab for the treatment of first-line non-squamous NSCLC, first-line OSCC or first-line G/GOJ adenocarcinoma, tumour PD-L1 status must be established. Testing used in clinical practice should be adequately comparable to the testing used in pivotal trials (see Section 5.1 Pharmacodynamic Properties, Clinical trials). A list of IVD companion diagnostic (CDx) tests is published on the TGA website.
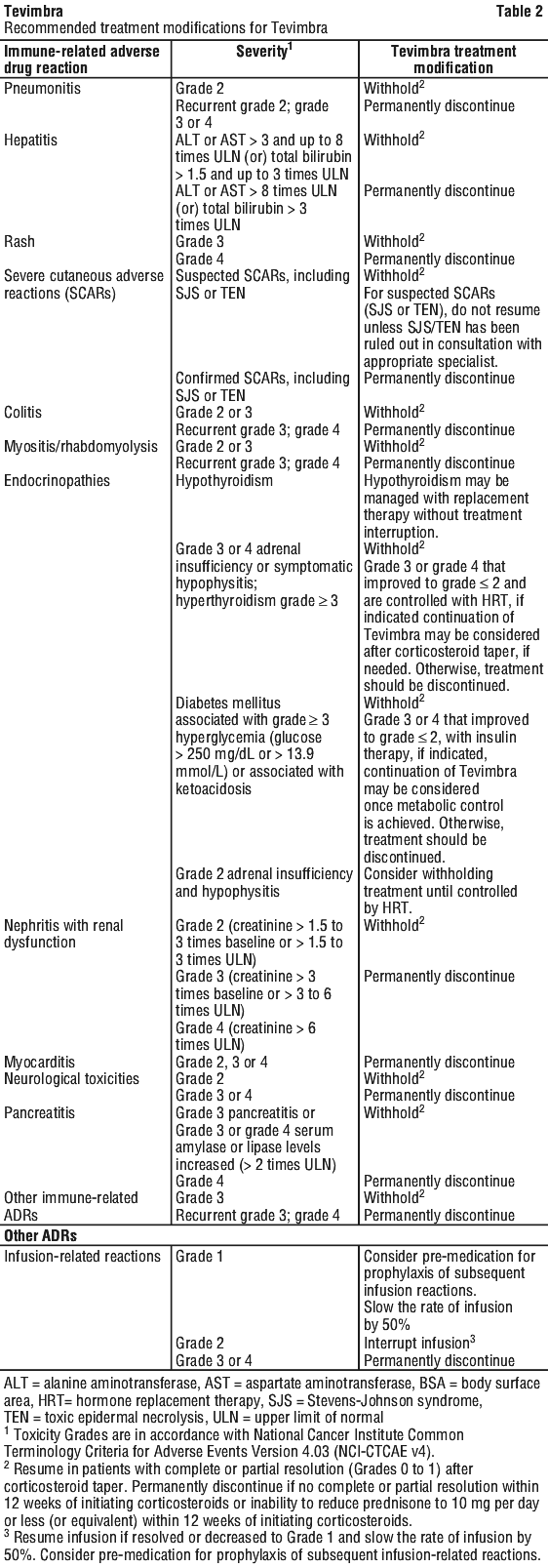
Treatment modifications for adverse drug reactions.
No dose reductions of Tevimbra as monotherapy or in combination therapy are recommended. Withhold or permanently discontinue Tevimbra depending on the severity of the adverse drug reaction (ADR).
Recommended treatment modifications to manage immune-related ADRs are provided in Table 2.
Detailed guidelines for the management of immune-related adverse reactions are described in Section 4.4 Special Warnings and Precautions for Use.
Special populations.
Patients with renal impairment.
Based on population pharmacokinetic analysis, no dose adjustment of Tevimbra is necessary in patients with mild or moderate renal impairment. Data from patients with severe renal impairment are too limited to draw conclusions for this population (see Section 5.2 Pharmacokinetic Properties).
Patients with hepatic impairment.
Based on population pharmacokinetic analysis, no dose adjustment of Tevimbra is necessary in patients with mild or moderate hepatic impairment. Data from patients with severe hepatic impairment are too limited to draw conclusions for this population (see Section 5.2 Pharmacokinetic Properties).
Use in children.
The efficacy and safety of Tevimbra has not been established in patients below 18 years.
Use in the elderly.
No dose adjustment of Tevimbra is required in patients ages 65 years or above (see Section 5.2 Pharmacokinetic Properties).
Method of administration.
Tevimbra is for intravenous infusion use only.
When Tevimbra is administered in combination with chemotherapy, it should be administered before chemotherapy when both are given on the same day. Refer to the Australian Prescribing Information for the chemotherapy administered in combination with Tevimbra.
Instructions for use and handling.
Vials are for single use only. Each vial contains 100 mg of tislelizumab.
The diluted solution for infusion should be prepared by a healthcare professional using aseptic technique.
Preparation of solution for infusion.
1. Remove the required number of vials from the refrigerator, taking care not to shake them.
2. Inspect each vial visually for particulate matter and discoloration prior to administration. The concentrate is a clear to slightly opalescent, colorless to slightly yellowish solution. Do not use a vial if the solution is cloudy or if visible particles or discoloration are observed.
3. Invert the vials gently, without shaking. Withdraw the required volume from the vial(s) and transfer into an intravenous (I.V.) infusion bag containing sodium chloride 9 mg/mL (0.9%) to prepare a diluted solution with a final concentration ranging from 2 to 5 mg/mL. Mix diluted solution by gentle inversion to avoid foaming or excessive shearing of the solution.
Administration.
1. Administer the diluted Tevimbra solution by I.V. infusion through an intravenous administration line with a sterile, non-pyrogenic, low-protein binding 0.2 micron or 0.22 micron in-line or add-on filter, with a surface area of approximately 10 cm2.
2. For 150 mg and 200 mg, the first infusion should be delivered over 60 minutes. If well tolerated, subsequent infusions may be administered over 30 minutes. For 300 mg, the first infusion should be delivered over 90 minutes. If this is well tolerated, the second infusion should be administered over 60 minutes. If the second infusion is well tolerated, subsequent infusions should be administered over 30 minutes.
3. Other drugs should not be co-administered through the same infusion line.
4. Tevimbra must not be administered as an intravenous push or single bolus injection.
5. Tevimbra does not contain any preservatives. It is recommended to prepare the solution immediately after taking it out of the refrigerator. From a microbiological point of view, once infusion is prepared, it is recommended to use the solution immediately after dilution. The diluted solution can be stored at 2°C to 8°C (36°F to 46°F) for up to 10 days (240 hours). The 10 days (240 hours) include storage of the diluted solution under refrigeration (2 to 8°C), and time required for returning to room temperature (25°C and below) as well as completing the infusion within 4 hours.
6. The diluted solution must not be frozen.
7. The intravenous line must be flushed at the end of the infusion.4.3 Contraindications
Hypersensitivity to tislelizumab or any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Patient card.
Patients treated with Tevimbra must be given a patient card to be informed about the risks of immune-related adverse reactions during tislelizumab therapy.
Prescribers must discuss the risks of immune-related adverse reactions during tislelizumab therapy with the patient.
Immune-related adverse drug reactions.
Severe, including fatal, cases of pneumonitis and hepatitis have been reported. Most immune-related adverse reactions occurring during treatment with tislelizumab were reversible and managed with interruptions of tislelizumab, administration of corticosteroids and/or supportive care. Immune-related adverse reactions have also occurred after the last dose of tislelizumab. Immune-related adverse reactions affecting more than one body system can occur simultaneously.
For suspected immune-related adverse reactions, adequate evaluation to confirm aetiology or exclude alternative aetiologies, including infection, should be ensured. Based on the severity of the adverse reaction, tislelizumab should be withheld and corticosteroids administered. Administration of other systemic immunosuppressants can be considered in patients whose immune-related adverse reactions are not controlled with corticosteroid therapy (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects)).
Patients with pre-existing autoimmune disease (AID). In patients with pre-existing autoimmune disease (AID), data from observational studies suggest that the risk of immune-mediated adverse reactions following immune checkpoint inhibitor therapy may be increased as compared with the risk in patients without pre-existing AID. In addition, flares of the underlying AID were frequent, but the majority were mild and manageable.
Immune-related pneumonitis. Immune-related pneumonitis, including fatal cases, has been reported in patients receiving Tevimbra. Patients should be monitored for signs and symptoms of pneumonitis. Patients with suspected pneumonitis should be evaluated with radiographic imaging and infectious or disease-related aetiologies should be excluded.
Patients with immune-related pneumonitis should be managed according to treatment modifications recommended in Table 2 (see Section 4.2 Dose and Method of Administration) as well as applicable current local treatment guidelines.
Immune-related hepatitis. Immune-related hepatitis has been reported in patients treated with Tevimbra, including fatal cases. Patients should be monitored for signs and symptoms of hepatitis and changes in liver function. Liver function tests (LFTs) should be performed at baseline and periodically during treatment.
Patients with immune-related hepatitis should be managed according to treatment modifications in Table 2 (see Section 4.2 Dose and Method of Administration) as well as applicable current local treatment guidelines.
Immune-related skin reactions. Cases of severe cutaneous adverse reactions (SCARs) including erythema multiforme (EM), Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), some with fatal outcome, have been reported in patients receiving Tevimbra (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be monitored for signs or symptoms of SCARs (e.g. a prodrome of fever, flu-like symptoms, mucosal lesions, or progressive skin rash) and other causes should be excluded. For suspected SCARs Tevimbra should be withheld, and the patient should be referred to specialised care for assessment and treatment. If SCARs is confirmed, Tevimbra should be permanently discontinued (see Section 4.2 Dose and Method of Administration).
Immune-related skin rash or dermatitis have been reported in patients receiving Tevimbra. Patients should be monitored for signs and symptoms of suspected skin reactions and other causes should be excluded. Based on the severity of the skin adverse reactions, Tevimbra should be withheld or permanently discontinued as recommended in Table 2 (see Section 4.2 Dose and Method of Administration) as well as per applicable current local treatment guidelines.
Immune-related colitis. Immune-related colitis, frequently associated with diarrhea, has been reported in patients treated with Tevimbra. Patients should be monitored for signs and symptoms of colitis. Infectious and disease-related aetiologies should be ruled out.
Patients with immune-related colitis should be managed according to treatment modifications in Table 2 (see Section 4.2 Dose and Method of Administration) as well as per applicable current local treatment guidelines.
Immune-related endocrinopathies. Immune-related endocrinopathies, including thyroid disorders, adrenal insufficiency, hypophysitis and diabetes mellitus, have been reported in patients treated with Tevimbra, which may require supportive treatment.
Patients with immune-related endocrinopathies should be managed according to treatment modifications as recommended in Table 2 (see Section 4.2 Dose and Method of Administration) as well as applicable local treatment guidelines.
Thyroid disorders.
Thyroid disorders, including hyperthyroidism, hypothyroidism and thyroiditis have been reported in patients treated with Tevimbra. Thyroiditis can present with or without other forms of endocrinopathy. Hypothyroidism can follow hyperthyroidism. Patients should be monitored for changes in thyroid function (at the start of treatment, periodically during treatment and as indicated based on clinical evaluation) and clinical signs and symptoms of thyroid disorders. Hypothyroidism may be managed with hormone replacement therapy without treatment interruption and without corticosteroids. Hyperthyroidism may be managed symptomatically.
Adrenal insufficiency.
Adrenal insufficiency has been reported in patients treated with Tevimbra. Patients should be monitored for signs and symptoms of adrenal insufficiency. Monitoring of adrenal function and hormone levels should be considered. Corticosteroids and hormone replacement should be administered as clinically indicated.
Hypophysitis/hypopituitarism.
Hypophysitis/hypopituitarism has been reported in patients treated with Tevimbra. Hypophysitis can cause hypopituitarism. Patients should be monitored for signs and symptoms of hypophysitis/hypopituitarism. Monitoring of pituitary function and hormone levels should be considered. Corticosteroids and hormone replacement should be administered as clinically indicated.
Diabetes mellitus.
Diabetes mellitus, including diabetic ketoacidosis has been reported in patients treated with Tevimbra. Patients should be monitored for hyperglycaemia or other signs and symptoms of diabetes. Insulin should be administered as clinically indicated for diabetes. In patients with severe hyperglycaemia or ketoacidosis (Grade ≥ 3), Tevimbra should be withheld and anti-hyperglycaemic treatment should be administered (see Section 4.2 Dose and Method of Administration). Treatment with Tevimbra should be resumed when metabolic control is achieved.
Immune-related nephritis with renal dysfunction. Immune-related nephritis with renal dysfunction has been reported in patients treated with Tevimbra. Patients should be monitored for changes in renal function (elevated serum creatinine) and other causes of renal dysfunction should be excluded.
Patients with immune-related nephritis with renal dysfunction should be managed according to treatment modifications in Table 2 (see Section 4.2 Dose and Method of Administration) as well as applicable current local treatment guidelines.
Other immune-related adverse reactions. Other clinically important immune-related adverse reactions were reported in patients treated with Tevimbra: myositis, myocarditis, arthritis, polymyalgia rheumatica, pericarditis, immune thrombocytopenia and Guillain-Barré syndrome (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients with other immune-related adverse reactions should be managed according to treatment modifications in Table 2 (see Section 4.2 Dose and Method of Administration) as well as applicable current local treatment guidelines.
Solid organ transplant rejection.
Solid organ transplant rejection has been reported in the post-marketing setting in patients treated with PD-1 inhibitors. Treatment with Tevimbra may increase the risk of rejection in solid organ transplant recipients. The benefit of treatment with Tevimbra versus the risk of possible organ rejection should be considered in these patients.
Haemaphagocytic lymphohistiocytosis (HLH).
Haemophagocytic lymphohistiocytosis (HLH) has been reported in patients receiving tislelizumab (see Section 4.8 Adverse Effects (Undesirable Effects)). HLH is a life-threatening syndrome characterised by fever, skin rash, lymphadenopathy, hepato- and/or splenomegaly and cytopenias. Patients should be monitored for clinical signs and symptoms of HLH. For suspected HLH, tislelizumab must be interrupted. If HLH is confirmed, administration of tislelizumab should be discontinued.
Infusion-related reactions.
Severe infusion-related reactions (Grade 3 or higher) have been reported in patients receiving Tevimbra (see Section 4.8 Adverse Effects (Undesirable Effects)). Cases of anaphylaxis, including anaphylactic reaction and anaphylactic shock, have been reported in the post-marketing setting. Patients should be monitored for signs and symptoms of infusion-related reactions.
Infusion-related reactions should be managed as recommended in Table 2 (see Section 4.2 Dose and Method of Administration).
Embryo-fetal toxicity.
There are no available data on the use of Tevimbra in pregnant women. Based on its mechanism of action, Tevimbra can cause fetal harm when administered to a pregnant woman.
Animal studies have demonstrated that inhibition of PD-1/PD-L1 pathway can lead to increased risk of immune-related rejection of the developing fetus resulting in fetal death.
Women should be advised of the potential risk to a fetus. Tevimbra should not be used during pregnancy and in women of childbearing potential not using effective contraception unless the clinical condition of the woman requires treatment with Tevimbra.
Sexually-active females of reproductive potential should be advised to use effective contraception (methods that result in less than 1% pregnancy rates) during treatment with Tevimbra and for at least 4 months after the last dose of Tevimbra (see Section 4.6 Fertility, Pregnancy and Lactation).
Patients on controlled sodium diet.
Each mL of this medicinal product contains 0.069 mmol (or 1.6 mg) sodium. This medicinal product contains 16 mg sodium per 10 mL vial, which is equivalent to 0.8% of the WHO recommended maximum daily intake of 2 g sodium for an adult.4.5 Interactions with Other Medicines and Other Forms of Interactions
Pharmacokinetic interaction studies have not been conducted. As monoclonal antibodies are not metabolised by cytochrome P450 (CYP) enzymes or other drug-metabolising enzymes, inhibition or induction of these enzymes by co-administered medicinal products is not anticipated to affect the pharmacokinetics of Tevimbra. Tevimbra is not expected to inhibit or induce CYP or other drug metabolising enzymes.
The use of systemic corticosteroids and other immunosuppressants at baseline, before starting Tevimbra, except for physiological doses of systemic corticosteroid (10 mg/day prednisone or equivalent), should be avoided because of their potential interference with the pharmacodynamic activity and efficacy. However, systemic corticosteroids and other immunosuppressants can be used after starting Tevimbra to treat immune-related adverse reactions (see Section 4.4 Special Warnings and Precautions for Use). When Tevimbra is administered in combination with chemotherapy, Tevimbra should be administered before chemotherapy when both are given on the same day. Refer to the prescribing information for the chemotherapy administered in combination with Tevimbra for recommendations on corticosteroid use as premedication for the prevention of chemotherapy-related adverse reactions.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No clinical data are available on the possible effects of tislelizumab on fertility. No reproductive and development toxicity studies have been conducted with tislelizumab.
(Category D)
There are no available data on the use of tislelizumab in pregnant women. Based on its mechanism of action, there is a potential risk that administration of tislelizumab during pregnancy may result in fetal harm.
Animal reproduction studies have not been conducted with tislelizumab. However, in murine models of pregnancy, blockade of PD1/PDL1 signaling has been shown to disrupt tolerance to the fetus and to result in increased fetal loss.
Human IgG4 (immunoglobulins) are known to cross the placental barrier. Therefore, tislelizumab, being an IgG4 variant, has the potential to be transmitted from the mother to the developing fetus. Tislelizumab is not recommended during pregnancy unless the clinical benefit is expected to outweigh the potential risk to the fetus. Tislelizumab should not be used during pregnancy and in women of childbearing potential not using effective contraception. Effective contraception (methods that result in less than 1% pregnancy rates) should be used for at least 4 months following the last dose of tislelizumab.
It is unknown whether tislelizumab is excreted in human milk but it is known that antibodies (including IgG4) are excreted in human milk. The effects of tislelizumab on breast-fed newborns/infants and on milk production are also unknown.
Because of the potential for serious adverse effects in breast-fed newborns/infants from tislelizumab, women should be advised not to breast-feed during treatment and for at least 4 months after the last dose of tislelizumab.4.7 Effects on Ability to Drive and Use Machines
Tevimbra has a minor influence on the ability to drive and use machines. In some patients, fatigue has been reported following administration of Tevimbra (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
Tislelizumab as monotherapy.
The safety of tislelizumab as monotherapy is based on pooled data in 2,390 patients across multiple tumour types. The most common adverse drug reaction (ADR) (reported at a frequency > 20%, with tislelizumab as monotherapy) were fatigue and increased aspartate aminotransferase. The most common Grade 3/4 ADRs (reported at a frequency > 2%, with tislelizumab as monotherapy) were increased aspartate aminotransferase, increased blood bilirubin and fatigue.
ADRs leading to death were reported in 0.3% of patients and include pneumonitis (0.1%), hepatitis (0.1%) and dyspnoea (0.04%).
Tislelizumab as combination therapy.
The safety of tislelizumab given in combination with chemotherapy is based on data in 1,950 patients treated with tislelizumab in combination with chemotherapy. Tislelizumab was administered at a dose of 200 mg every 3 weeks in combination with chemotherapy. The most common ADRs (reported at a frequency > 20%, with tislelizumab in combination with chemotherapy) were, fatigue, increased alanine aminotransferase, increased aspartate aminotransferase, diarrhoea and rash. The most common Grade 3/4 ADRs (reported at a frequency > 2%, with tislelizumab in combination with chemotherapy) were fatigue, hypokalaemia, rash, increased alanine aminotransferase and increased aspartate aminotransferase.
ADRs leading to death were reported in 0.8% of patients. The ADRs leading to death were pneumonitis (0.3%), myocarditis (0.2%), dyspnoea (0.2%), hepatitis (0.1%), colitis (0.1%), hypokalaemia (0.1%), and myositis (0.1%).
Tabulated list of adverse reactions.
Table 3 lists the incidence of adverse reactions in the monotherapy safety dataset and in patients treated with tislelizumab in combination with chemotherapy. ADRs are listed according to system organ class in MedDRA. Within each system organ class, the adverse drug reactions are presented in decreasing frequency.
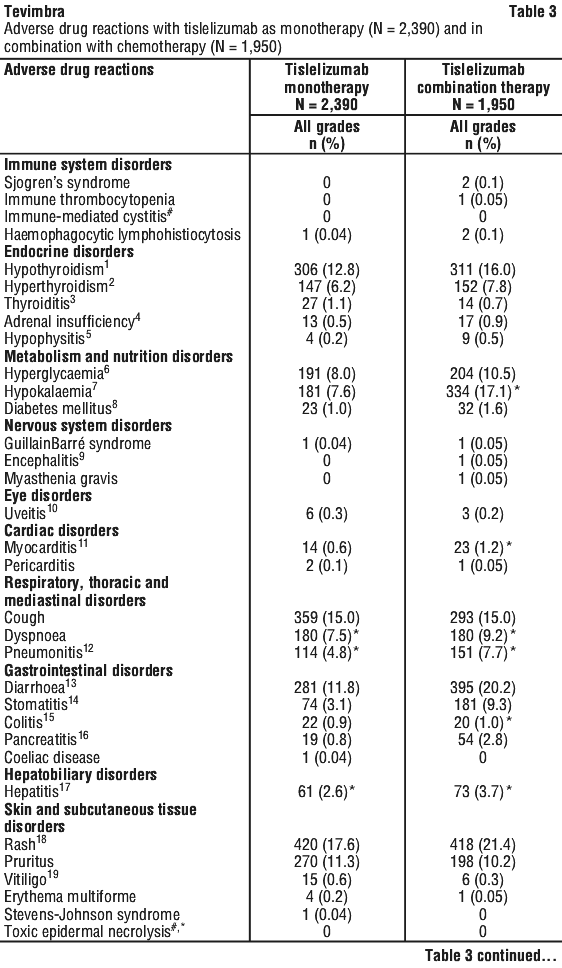

Adverse events by indication.
Oesophageal squamous cell carcinoma (OSCC). First-line treatment of unresectable, locally advanced, or metastatic oesophageal carcinoma.
The safety of Tevimbra in combination with chemotherapy was evaluated in RATIONALE-306, a randomised, placebo-controlled, multicenter, double-blind trial in patients with unresectable, locally advanced, or metastatic OSCC.
Patients were randomised (1:1) to receive either Tevimbra 200 mg or placebo plus a chemotherapy doublet regimen. The chemotherapy doublet regimens consist of:
platinum (cisplatin [60 to 80 mg/m2 IV, on Day 1] or oxaliplatin [130 mg/m2 IV, on Day 1]) and a fluoropyrimidine (5-FU [750 to 800 mg/m2 IV, on Day 1 to 5] or capecitabine [1000 mg/m2 orally twice daily, on Day 1 to 14]); or
platinum (cisplatin [60 to 80 mg/m2 IV, on Day 1 or 2] or oxaliplatin [130 mg/m2 IV, on Day 1 or 2]) and (paclitaxel 175 mg/m2 IV, on Day 1).
The median tislelizumab/placebo duration of exposure was 6.41 months (range, 0.1 to 38.3 months) in the Tevimbra plus chemotherapy arm.
The most common adverse events (≥ 30%) were anaemia, neutrophil count decreased, decreased appetite, white blood cell count decreased, nausea, and constipation.
The most common ≥ Grade 3 adverse events (≥ 5%) were neutrophil count decreased, anaemia, hyponatraemia, white blood cell count decreased, hypokalaemia, neutropenia, dysphagia, pneumonia, decreased appetite, and fatigue.
Serious adverse events occurred in 48.1% of patients; the most frequent serious adverse events (≥ 2%) were dysphagia, pneumonia, diarrhoea, and oesophageal stenosis.
Fatal adverse events occurred in 5.2% of patients who received tislelizumab, including acquired trachea-oesophageal fistula, brain injury, electrolyte imbalance, gastrointestinal haemorrhage, multiple organ dysfunction syndrome, myocarditis, pneumonia, pneumonia aspiration, pulmonary embolism, pulmonary tuberculosis, respiratory failure, sepsis, sudden death, and traumatic intracranial haemorrhage (1 patient each), and death (2 patients).
Adverse events leading to discontinuation of tislelizumab occurred in 13.0% of patients; the most common adverse events resulting in permanent discontinuation (≥ 1%) was pneumonitis.
Adverse events leading to the treatment modification of tislelizumab occurred in 52.5% of patients; the most common adverse events leading to treatment modification of tislelizumab (≥ 5%) were neutrophil count decreased and pneumonia.
Adverse events are listed in Table 4.
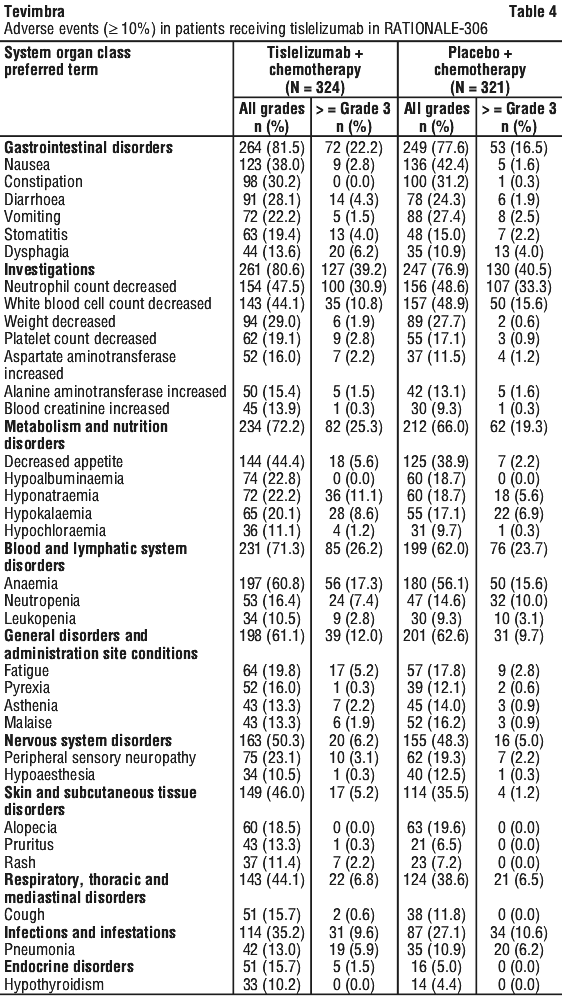
Second-line treatment of unresectable, locally advanced, or metastatic oesophageal carcinoma.
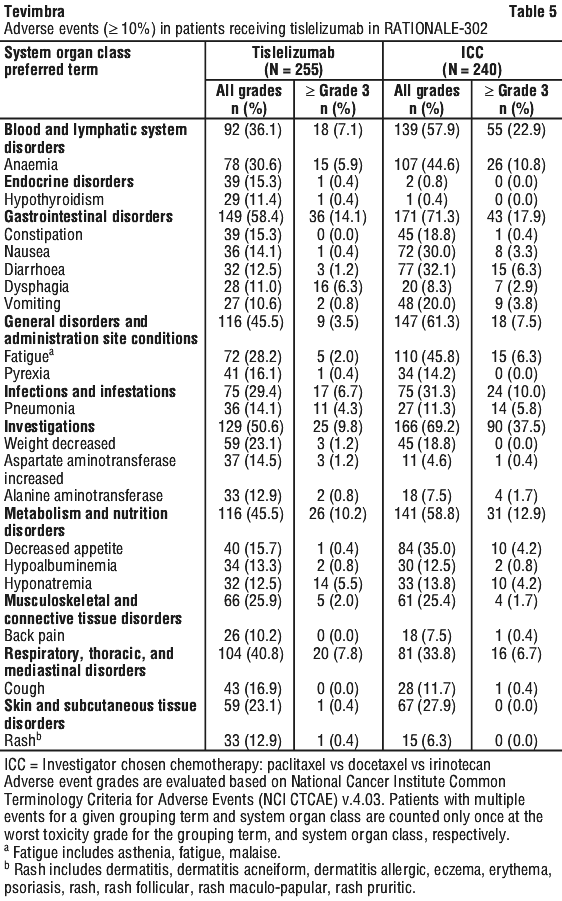
The data described below reflect exposure to tislelizumab in 255 patients with oesophageal squamous cell carcinoma in RATIONALE-302. Patients received 200 mg of tislelizumab via intravenous infusion every 3 weeks [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. The median duration of exposure to tislelizumab was 2.8 months (range: 0.2 to 28.3 months).
The most common adverse events (≥ 20%) were anaemia, fatigue (including asthenia, fatigue, and malaise), and weight decreased.
The most common ≥ Grade 3 adverse events (≥ 2%) were dysphagia, anaemia, hyponatremia, pneumonia, dyspnoea, lymphocyte count decreased, hypertension, fatigue (including asthenia, fatigue, and malaise), and pneumonitis (including pneumonitis and interstitial lung disease).
Serious adverse events occurred in 41.2% of patients; the most frequent serious adverse events (≥ 2%) were pneumonia, dysphagia, pneumonitis (including pneumonitis and immune-mediated pneumonitis), and oesophageal obstruction.
Fatal adverse events (excluding death due to disease under study) occurred in 5.5% of patients who received tislelizumab, including bronchiectasis, hemoptysis, pulmonary arterial hypertension, pulmonary embolism, pulmonary hemorrhage, sudden death, cardio-respiratory arrest, upper gastrointestinal hemorrhage, platelet count decreased (1 patient each), death due to an unknown cause (2 patients), and pneumonia (3 patients).
Adverse events leading to discontinuation of tislelizumab occurred in 19.2% of patients; the most common adverse events resulting in permanent discontinuation (≥ 1%) were pneumonitis (including pneumonitis and immune-mediated pneumonitis), pneumonia, and upper gastrointestinal hemorrhage.
Adverse events leading to the interruption of tislelizumab occurred in 22.7% of patients; the most common adverse events leading to interruption of tislelizumab (≥ 2%) were pneumonia, pneumonitis (including pneumonitis, interstitial lung disease, and immune-mediated pneumonitis) and fatigue (including asthenia and fatigue).
Adverse events are listed in Table 5.
 Non-small cell lung cancer (NSCLC).
Non-small cell lung cancer (NSCLC). First-line treatment of metastatic non-squamous NSCLC in combination with pemetrexed and platinum chemotherapy.
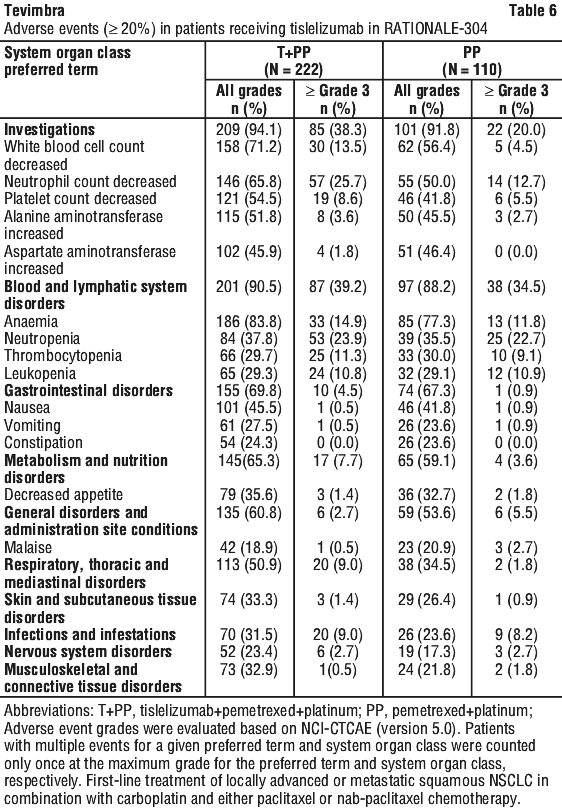
The data described below reflect exposure to tislelizumab in 222 patients with untreated locally advanced or metastatic non-squamous NSCLC in RATIONALE-304. Patients received tislelizumab 200 mg combined with pemetrexed 500 mg/m2 and carboplatin AUC 5 mg/mL/min or cisplatin 75 mg/m2 (T+PP arm) [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. The median duration of exposure to tislelizumab was 34.14 weeks (range: 3.0 to 118.0 weeks).
The most common adverse events (≥ 50%) were anaemia, white blood cell count decreased, neutrophil count decreased, platelet count decreased, and alanine aminotransferase increased.
The most common ≥ Grade 3 adverse events (≥ 5%) were neutropenia, anaemia, thrombocytopenia, leukopenia, neutrophil count decreased, white blood cell count decreased, platelet count decreased, and pneumonia.
Serious adverse events occurred in 39.2% of patients; the most frequent serious adverse events (≥ 2%) were pneumonitis, pneumonia, thrombocytopenia, platelet count decreased, pyrexia, and dyspnoea.
Fatal adverse events occurred in 4.1% of patients who received tislelizumab, including asphyxia, dyspnoea, atrial fibrillation, myocarditis, death, and cerebellar haemorrhage (1 patient each), and pneumonitis (3 patients).
Adverse events leading to discontinuation of tislelizumab occurred in 14.4% of patients; the most common adverse events resulting in permanent discontinuation (≥ 1%) were pneumonitis, and immune-mediated enterocolitis.
Adverse events leading to the treatment modification of tislelizumab occurred in 64.0% of patients; the most common adverse events leading to treatment modification of tislelizumab (≥ 5%) were anaemia, neutropenia, white blood cell count decreased, neutrophil count decreased, alanine aminotransferase increased, pneumonia, and pneumonitis.
Adverse events are listed in Table 6.
First-line treatment of metastatic squamous NSCLC in combination with paclitaxel and platinum chemotherapy.
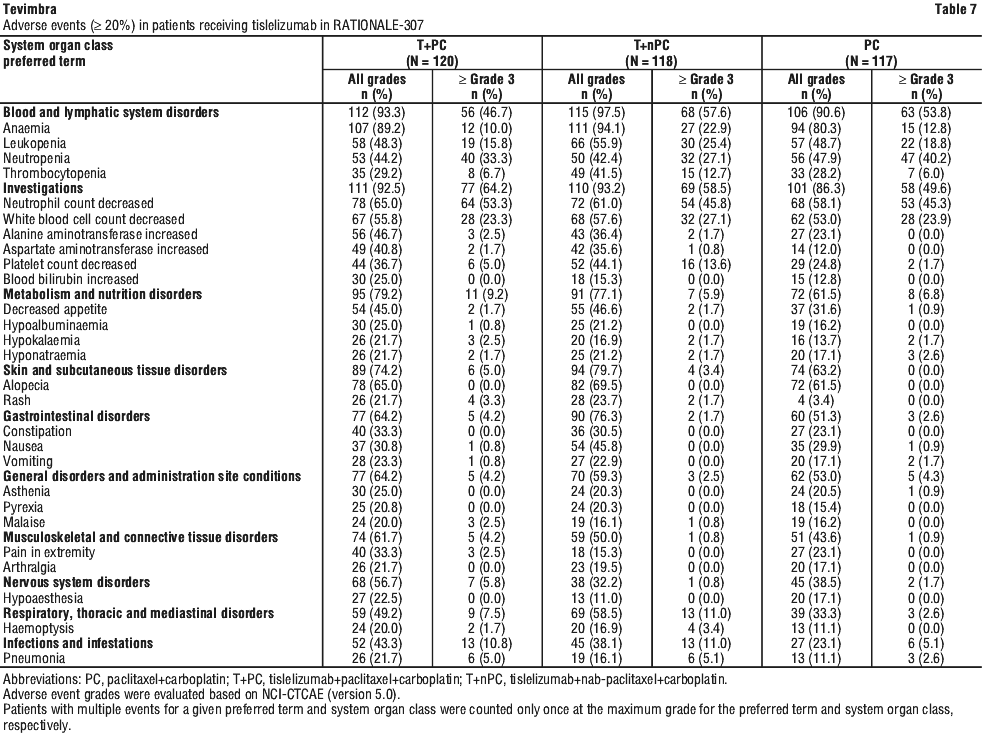
The data described below reflect exposure to tislelizumab in 238 patients with locally advanced or metastatic squamous NSCLC in RATIONALE-307. Patients receive tislelizumab 200 mg combined with paclitaxel 175 mg/m2 and carboplatin AUC 5 mg/mL/min (T+PC arm, N = 120), or tislelizumab 200 mg combined with nab-paclitaxel 100 mg/m2 and carboplatin AUC 5 mg/mL/min (T+nPC arm, N = 118) [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. The median duration of exposure to tislelizumab was 40.21 weeks (range: 3.0 to 100.9 weeks) in Arm T+PC and 44.21 weeks (range: 3.0 to 105.0 weeks) in Arm T+nPC.
The most common adverse events (≥ 50%) were anaemia, neutrophil count decreased, white blood cell count decreased, and alopecia in Arm T+PC, and anaemia, leukopenia, neutrophil count decreased, white blood cell count decreased, and alopecia in Arm T+nPC.
The most common ≥ Grade 3 adverse events (≥ 5%) were neutrophil count decreased, white blood cell count decreased, platelet count decreased, neutropenia, anaemia, thrombocytopenia, leukopenia, and pneumonia in both Arm T+PC and Arm T+nPC.
Serious adverse events occurred in 43.3% of patients in Arm T+PC and 42.4% of patients in Arm T+nPC. The most frequent serious adverse events (≥ 2%) were pneumonitis, pneumonia, haemoptysis, and neutrophil count decreased in Arm T+PC, and pneumonitis, pneumonia, haemoptysis, febrile neutropenia, and neutrophil count decreased in Arm T+nPC.
Fatal adverse events occurred in 3.3% of patients in Arm T+PC and 5.9% of patients in Arm T+nPC, including cerebrovascular accident, hydrocephalus, haemoptysis, and respiratory failure (1 patient each) in Arm T+PC, and haemoptysis, respiratory failure, hepatic failure, pneumonia, and hypokalaemia (1 patient each) and death (2 patients) in Arm T+nPC.
Adverse events leading to discontinuation of tislelizumab occurred in 14.2% of patients in Arm T+PC and 12.7% of patients in Arm T+nPC; the most common adverse events resulting in permanent discontinuation (≥ 2 patients) were pneumonitis, and pneumonia in Arm T+PC, and immune-mediated pneumonitis, pneumonia, blood creatine phosphokinase increased, myocarditis, and death in Arm T+nPC.
Adverse events leading to the treatment modification of tislelizumab occurred in 47.5% of patients in Arm T+PC, and 79.7% of patients in Arm T+nPC; the most common adverse events leading to treatment modification of tislelizumab (≥ 5%) were anaemia, thrombocytopenia, leukopenia, platelet count decreased, neutrophil count decreased, alanine aminotransferase increased, white blood cell count decreased, aspartate aminotransferase increased, hypothyroidism, and pneumonia in Arm T+PC, and anaemia, thrombocytopenia, leukopenia, neutropenia, platelet count decreased, neutrophil count decreased, alanine aminotransferase increased, and white blood cell count decreased in Arm T+nPC.
Adverse events are listed in Table 7.
Previously treated non-small cell lung cancer.
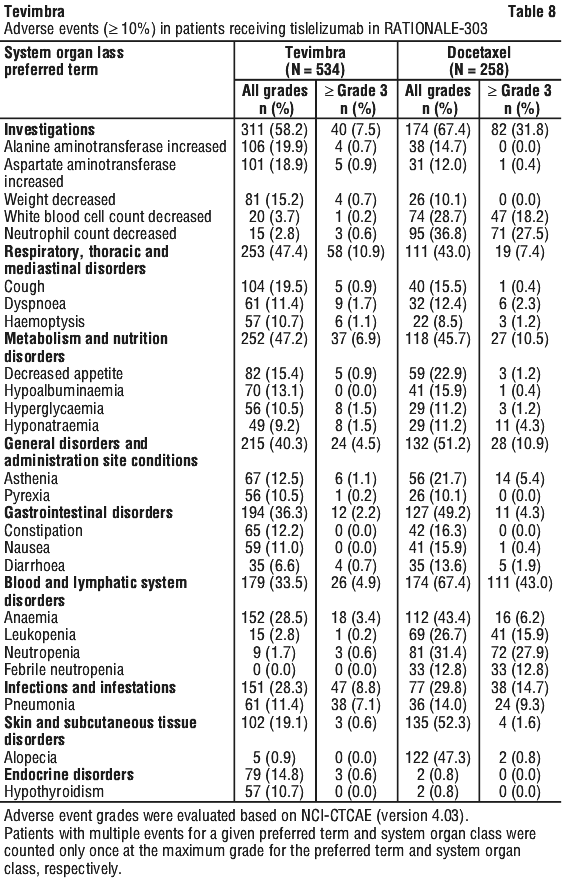
The data described below reflect exposure to tislelizumab in 534 patients with locally advanced or metastatic NSCLC (squamous or non-squamous) in RATIONALE-303. Patients received 200 mg of Tevimbra via intravenous infusion every 3 weeks [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. The median duration of exposure to tislelizumab was 23.3 weeks (range: 1 to 140 weeks).
The most common adverse events (≥ 15%) were anaemia, decreased appetite, cough, weight decreased, alanine aminotransferase increased, and aspartate aminotransferase increase.
The most common ≥ Grade 3 adverse events (≥ 5%) were pneumonia.
Serious adverse events occurred in 32.6% of patients; the most frequent serious adverse events (≥ 1%) were pneumonia, pneumonitis, immune-mediated pneumonitis, interstitial lung disease, haemoptysis, dyspnoea, and pleural effusion.
Fatal adverse events related to tislelizumab occurred in 1.5% of patients, including multiple organ dysfunction syndrome, pneumonitis, and hepatic function abnormal (1 patient each), death, respiratory failure, and pneumonia (2 patients each).
Adverse events leading to discontinuation of tislelizumab occurred in 10.5% of patients; the most common adverse events resulting in permanent discontinuation (≥ 1%) were pneumonitis, interstitial lung disease and pneumonia.
Adverse events leading to the interruption of tislelizumab occurred in 22.3% of patients; the most common adverse events leading to interruption of tislelizumab (≥ 2%) were pneumonia, and blood creatine phosphokinase increased.
Adverse events are listed in Table 8.
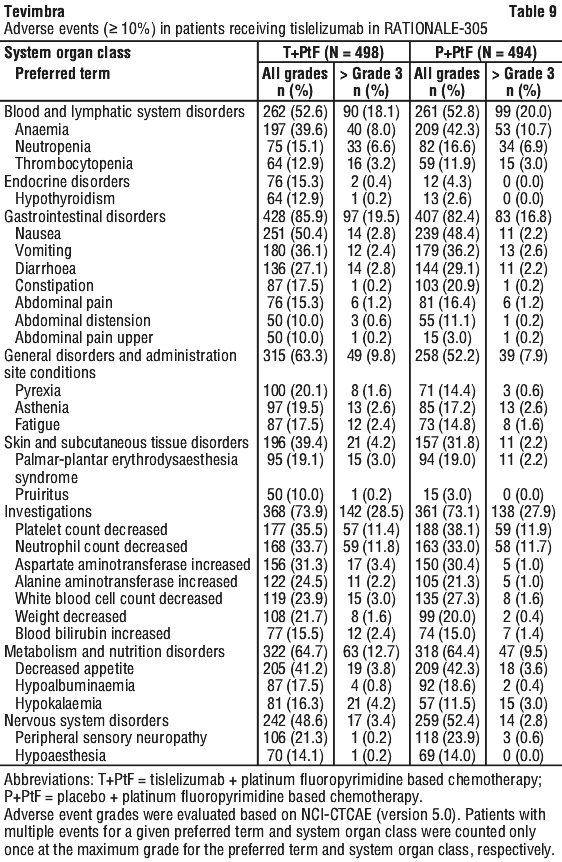
 Gastric/gastro-oesophageal junction (G/GOJ) adenocarcinoma. The data described below reflect exposure to tislelizumab in 498 patients with previously untreated locally advanced unresectable or metastatic gastric or gastro-oesophageal (G/GOJ) junction adenocarcinoma in RATIONALE-305. Patients received tislelizumab 200 mg plus platinum and fluoropyrimidine based chemotherapy (T+PtF arm) (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The median duration of exposure to tislelizumab was 25.7 weeks (range: 0.4 to 204.4 weeks).
Gastric/gastro-oesophageal junction (G/GOJ) adenocarcinoma. The data described below reflect exposure to tislelizumab in 498 patients with previously untreated locally advanced unresectable or metastatic gastric or gastro-oesophageal (G/GOJ) junction adenocarcinoma in RATIONALE-305. Patients received tislelizumab 200 mg plus platinum and fluoropyrimidine based chemotherapy (T+PtF arm) (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The median duration of exposure to tislelizumab was 25.7 weeks (range: 0.4 to 204.4 weeks).
The most common adverse events (≥ 30%) were nausea, decreased appetite, anaemia, vomiting, platelet count decreased, neutrophil count decreased and aspartate aminotransferase increased.
The most common ≥ Grade 3 adverse events (≥ 5%) were neutropenia, neutrophil count decreased, platelet count decreased and anaemia.
Serious adverse events occurred in 42.4% of patients who received tislelizumab; the most frequent serious adverse events (≥ 2%) were platelet count decreased, pneumonia and death.
Fatal adverse events occurred in 9.4% of patients. Excluding cases where the primary reason for death was disease progression, death due to adverse event occurred in 4.2% of patients, including death, sepsis, pulmonary embolism, respiratory failure, colitis, pneumonia, pneumonia bacterial, respiratory tract infection viral, shock hemorrhagic, subdural hematoma and sudden death.
Adverse events leading to discontinuation of tislelizumab occurred in 15.7% of patients; the most common adverse events resulting in permanent discontinuation (≥ 1%) were death, peripheral sensory neuropathy, platelet count decreased and pneumonitis.
Adverse events leading to the treatment modification of tislelizumab occurred in 49.0% of patients; the most common adverse events leading to treatment modification of tislelizumab (≥ 10%) were platelet count decreased, neutrophil count decreased and white blood cell count decreased.
Adverse events are listed in Table 9.
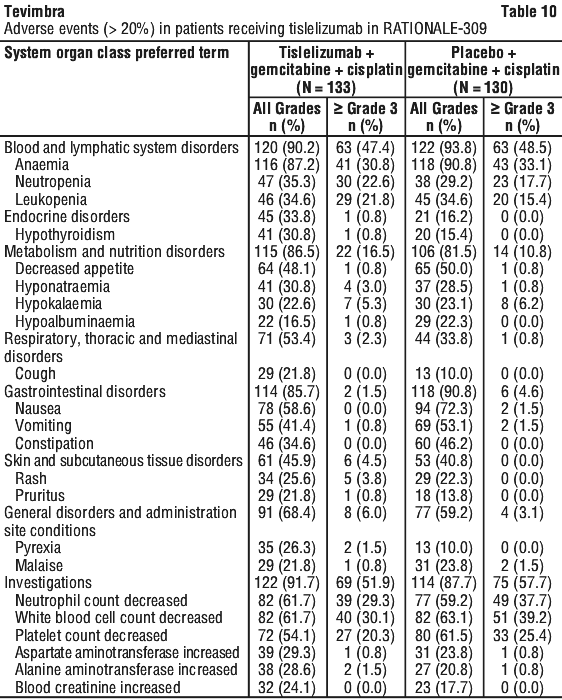
 Nasopharyngeal carcinoma (NPC). The data described below reflect exposure to tislelizumab in 263 patients with recurrent or metastatic NPC in RATIONALE-309. Patients received tislelizumab 200 mg once every 3 weeks in combination with gemcitabine plus cisplatin (T+GC arm) [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. The median duration of exposure to tislelizumab was 10.51 months (range 0.3 to 53.1 months).
Nasopharyngeal carcinoma (NPC). The data described below reflect exposure to tislelizumab in 263 patients with recurrent or metastatic NPC in RATIONALE-309. Patients received tislelizumab 200 mg once every 3 weeks in combination with gemcitabine plus cisplatin (T+GC arm) [see Section 5.1 Pharmacodynamic Properties, Clinical trials]. The median duration of exposure to tislelizumab was 10.51 months (range 0.3 to 53.1 months).
The most common adverse events (≥ 50%) were decreased white blood cell count, decreased neutrophil count, nausea, decreased platelet count, decreased appetite and vomiting.
The most common ≥ Grade 3 adverse events (≥ 30%) were anaemia and decreased white blood cell count.
Serious adverse events occurred in 35.3% of patients who received tislelizumab; the most frequent serious adverse event (≥ 5%) was decreased platelet count.
Fatal adverse events occurred in 4.5% of patients. The cause of death for 2 patients in the T+GC arm was disease under study, while the primary cause of death for the remaining 4 patients was due to adverse events, including pneumonia aspiration and hypoxia (1 patient), myelodysplastic syndrome, death, and accidental death (1 patient each).
Adverse events leading to discontinuation of tislelizumab occurred in 16.5% of patients; the most common adverse events resulting in permanent discontinuation (≥ 1%) were leukopenia, myocarditis, decreased neutrophil count, decreased platelet count, thrombocytopenia and decreased white blood cell count.
Adverse events leading to the treatment modification of tislelizumab occurred in 72.9% of patients; the most common adverse events leading to treatment modification of tislelizumab (≥ 10%) were decreased white blood cell count, decreased neutrophil count, neutropenia, leukopenia, decreased platelet count and anaemia.
Adverse events are listed in Table 10.
Laboratory abnormalities.
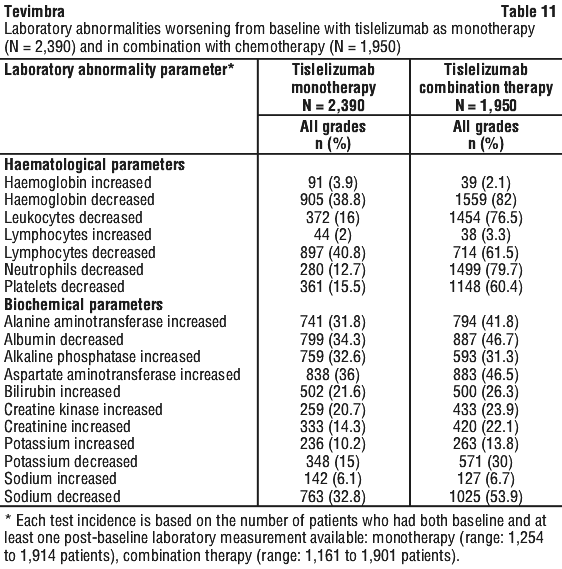
Clinically relevant abnormalities of routine haematological or biochemical laboratory values from the data set of the 9 pooled studies of tislelizumab as monotherapy and the 9 pooled studies of tislelizumab in combination with chemotherapy are presented in Table 11.
Description of selected adverse drug reactions.
Immune-related ADRs. The data below reflect information for ADRs for tislelizumab as monotherapy in clinical studies. Details for the ADRs for Tevimbra when given in combination are presented if clinically relevant differences were noted in comparison to Tevimbra monotherapy.
Immune-related pneumonitis. In patients treated with Tevimbra as monotherapy, immune-related pneumonitis occurred in 113 (4.7%) of 2,390 patients, including Grade 1 (25 patients, 1.0%), Grade 2 (46 patients, 1.9%), Grade 3 (33 patients, 1.4%), Grade 4 (6 patients, 0.3%) and Grade 5 (3 patients, 0.1%) events.
The median time from first dose to onset of the event was 3.9 months (range: 1.0 day to 55 months), and the median duration of the event was 6 months (range: 1+ day to 48.3+ months). + denotes a censored observation, with ongoing events at the time of the analysis. Tevimbra was permanently discontinued in 44 (1.8%) patients and Tevimbra treatment was interrupted in 40 (1.7%) patients.
Eighty-one (71.7%) of the 113 patients received systemic corticosteroids. + denotes a censored observation. 74 (65.5%) of the 113 patients received high-dose (defined as a dose ≥ 40 mg/day of prednisone or equivalent) systemic corticosteroids. Two (1.8%) of 113 patients received immunosuppressive treatment. Pneumonitis resolved in 55 (48.7%) of the 113 patients.
Immune-related hepatitis. In patients treated with tislelizumab as monotherapy, immune-related hepatitis occurred in 30 (1.3%) of 12,390 patients, including Grade 1 (2 patients, 0.1%), Grade 2 (7 patients, 0.3%), Grade 3 (15 patients, 0.6%) and Grade 4 (4 patients, 0.3%).
The median time from first dose to onset of the event was 1.1 months (range: 14 days to 34.8 months), and the median duration of the event was 1.9 months (range: 6 days to 6.6 months). Tislelizumab was permanently discontinued in 6 (0.3%) patients and tislelizumab treatment was interrupted in 19 (0.8%) patients for immune-related hepatitis.
Twenty-five (83.3%) out of 30 patients received systemic corticosteroids. Twenty-four (80%) of the 30 patients received high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). Two (6.7%) of the 30 patients received other immunosuppressive treatment. Hepatitis resolved in 20 (66.7%) of the 30 patients. The median duration for all resolved events was 27.5 days (range: 6 days to 6.6 months).
Immune-related skin adverse reactions. In patients treated with tislelizumab as monotherapy, immune-related skin adverse reactions occurred in 311 (13%) of 2,390 patients, including Grade 1 (201 patients, 8.4%), Grade 2 (82 patients, 3.4%), Grade 3 (26 patients, 1.1%) and Grade 4 (2 patients, 0.1%) events.
The median time from first dose to onset of the event was 1.5 months (range: 1.0 day to 36.1 months). The median duration of the event was 2.1 months (range: 1.0 day to 61.6+ months). + denotes a censored observation. Tislelizumab was permanently discontinued in 3 (0.1%) patients, and tislelizumab treatment was interrupted in 30 (1.3%) patients.
Forty-four (14.1%) of the 311 patients received systemic corticosteroids. Nineteen (6.1%) of the 311 patients received high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). Two out of 311 patients (0.6%) received immunosuppressive treatment. Skin adverse reactions resolved in 208 (66.9%) of the 311 patients. The median duration for all resolved events was 32 days (range: 1 day to 36.7 months).
Immune-related colitis. In patients treated with tislelizumab as monotherapy, immune-related colitis occurred in 19 (0.8%) of 2,390 patients, including Grade 1 (1 patient, 0.04%), Grade 2 (10 patients, 0.4%) and Grade 3 (8 patients, 0.3%) events.
The median time from first dose to onset of the event was 6 months (range: 6 days to 26.5 months), and the median duration of the event was 26 days (range: 5 days to 26.7 months). Tislelizumab was permanently discontinued in 5 (0.2%) patients and tislelizumab treatment was interrupted in 10 (0.4%) patients.
Seventeen of the 19 patients received systemic corticosteroids. Twelve (63.2%) of the 19 patients were treated with high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). Two (10.5%) of the 19 patients received immunosuppressive treatment. Colitis resolved in 17 (89.5%) of the 19 patients. The median duration for all resolved events was 26 days (range: 5 days to 26.7 months).
Immune-related myositis/rhabdomyolysis. In patients treated with tislelizumab as monotherapy, immune-related myositis/rhabdomyolysis occurred in 17 (0.7%) of 2,390 patients, including Grade 1 (7 patients, 0.3%), Grade 2 (5 patients, 0.2%), Grade 3 (4 patients, 0.2%) and Grade 4 (1 patient, 0.04%) events.
The median time from first dose to onset of the event was 1.6 months (range: 15 days to 39.3 months), and the median duration of the event was 43 days (range: 5 days to 5.2 months). Tislelizumab was permanently discontinued in 4 (0.2%) patients and tislelizumab treatment was interrupted in 9 (0.4%) patients.
Nine (52.9%) of the 17 patients received systemic corticosteroids. Eight (47.1%) of the 17 patients were treated with high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). One (5.9%) out of the 17 patients received immunosuppressive treatment. Myositis/rhabdomyolysis resolved in 13 (76.5%) of the 17 patients. The median duration for all resolved events was 40.5 days (range: 5 days to 5.2 months).
Immune-related endocrinopathies. Thyroid disorders.
Hypothyroidism: in patients treated with tislelizumab as monotherapy, hypothyroidism occurred in 299 (12.5%) of 2,390 patients, including Grade 1 (137 patients, 5.7%), Grade 2 (160 patients, 6.7%), Grade 3 (1 patient, 0.04%), and Grade 4 (1 patient, 0.04%) events.
The median time from first dose to onset of the event was 3.7 months (range: 1 day to 29.9 months). The median duration of the event was 10.3 months (range: 1+ day to 56+ months). + denotes a censored observation. Tislelizumab was permanently discontinued in 2 (0.1%) patients and tislelizumab treatment was interrupted in 12 (0.5%) patients.
Two (0.7%) of the 299 patients received systemic corticosteroids. No patient received high-dose systemic corticosteroids. One hundred ninety-five patients received hormone replacement therapy. Hypothyroidism resolved in 103 (34.4%) of the 299 patients. The median duration for all resolved events was 2.1 months (range: 2 days to 27 months).
Hyperthyroidism: in patients treated with tislelizumab as monotherapy, hyperthyroidism occurred in 118 (4.9%) of 2,390 patients, including Grade 1 (95 patients, 4.0%), Grade 2 (22 patients, 0.9%) and Grade 3 (1 patient, 0.04%) events.
The median time from first dose to onset of the event was 2.1 months (range: 6 days to 39.4 months). The median duration of the event was 2.1 months (range: 8 days to 48.4+ months). + denotes a censored observation. Tislelizumab was permanently discontinued in one (0.04%) patient and tislelizumab treatment was interrupted in 7 (0.3%) patients.
Two (2.5%) of the 118 patients received systemic corticosteroids. One patient received high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). Nineteen (16.1%) of the 118 patients received hormone replacement therapy. Hyperthyroidism resolved in 90 (76.3%) of the 118 patients. The median duration for all resolved events was 1.4 months (range: 8 days to 22.1 months).
Thyroiditis: in patients treated with tislelizumab as monotherapy, thyroiditis occurred in 25 (1.0%) of 2,390 patients, including Grade 1 (124 patients, 0.5%) and Grade 2 (13 patients, 0.5%) events.
The median time from first dose to onset of the event was 2.0 months (range: 14 days to 20.7 months). The median duration of the event was 7.3 months (range: 20 days to 34.5+ months). + denotes a censored observation. Tislelizumab was not permanently discontinued in any patient and tislelizumab treatment was interrupted in 5 (0.2%) patients.
Two (8%) of the 25 patients received systemic corticosteroids. No patients received high-dose systemic corticosteroids. Sixteen (64%) of the 25 patients received hormone replacement therapy. Thyroiditis resolved in 9 (36%) of the 25 patients. The median duration for all resolved events was 2 months (range: 20 days to 15.3 months).
Adrenal insufficiency.
In patients treated with tislelizumab as monotherapy, adrenal insufficiency occurred in 12 (0.5%) of 2,390 patients, including Grade 2 (7 patients, 0.3%), Grade 3 (4 patients, 0.2%) and Grade 4 (1 patient, 0.04%) events.
The median time from first dose to onset of the event was 9.7 months (range: 1.4 months to 16.9 months). The median duration of the event was not reached (range: 1 month to 27.9+ months). + denotes a censored observation. Tislelizumab was not permanently discontinued in any patient and tislelizumab treatment was interrupted in 10 (0.4%) patients.
All 12 patients received systemic corticosteroids. Three (25%) of the 12 patients received high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). Adrenal insufficiency resolved in 3 (25%) of the 12 patients. The median duration for all resolved events was 1.9 months (range: 30 days to 13.6 months).
Hypophysitis.
In patients treated with tislelizumab as monotherapy, hypophysitis occurred in 6 (0.3%) of 2,390 patients, all Grade 2.
The median time from first dose to onset of the event was 8.7 months (range: 22 days to 16.2 months). The median duration of the event was not reached (range: 70 days to 30+ months). + denotes a censored observation. Tislelizumab was not permanently discontinued in any patients and treatment was interrupted in 1 (0.04%) patient.
Five (83.3%) of the 6 patients received systemic corticosteroids. One (16.7%) of the 6 patients received high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). Hypophysitis resolved in 1 (16.7%) of the 6 patients. The duration for the resolved event was 2.3 months.
Diabetes mellitus.
In patients treated with tislelizumab as monotherapy, diabetes mellitus occurred in 16 (0.7%) patients, including Grade 1 (1 patient, 0.04%), Grade 2 (6 patients, 0.3%), Grade 3 (6 patients, 0.3%) and Grade 4 (3 patients, 0.1%) events.
The median time from first dose to onset of the event was 6.5 months (range: 22 days to 36.1 months). The median duration of the event was not reached (range: 2 days to 44.5 months). + denotes a censored observation. Tislelizumab was permanently discontinued in 0.2% of patients and tislelizumab treatment was interrupted in 0.2% of patients. Diabetes mellitus resolved in two (12.5%) of 16 patients. The median duration for all resolved events was 22 days (range: 2 days to 3.6 months). Fourteen (87.5%) of the 16 patients received insulin therapy for diabetes mellitus.
Immune-related nephritis and renal dysfunction. In patients treated with tislelizumab as monotherapy, immune-related nephritis and renal dysfunction occurred in 5 (0.2%) of 2,390 patients, including Grade 1 (1 patient, 0.04%), Grade 2 (3 patients, 0.1%), Grade 3 (1 patient, 0.04%).
The median time from first dose to onset of the event was 2.1 months (range: 15 days to 34.5 months). The median duration of the event was not reached (range: 9 days to 16.2+ months). + denotes a censored observation. Tislelizumab was permanently discontinued in 1 (0.04%) patient and tislelizumab treatment was interrupted in 3 (0.1%) patients.
Three (60%) out of 5 patients received systemic corticosteroids. All 3 patients received high-dose systemic corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). One (20%) of the 5 patients received immunosuppressive treatment. Immune-related nephritis and renal dysfunction resolved in 2 (40%) of the 5 patients. The duration for all resolved events was 9 days.
Immune-related myocarditis. In patients treated with tislelizumab as monotherapy, immune-related myocarditis occurred in 15 (0.6%) of 2,390 patients, including Grade 1 (8 patients, 0.3%), Grade 2 (3 patients, 0.1%), Grade 3 (3 patients, 0.1%) and Grade 4 (1 patient, 0.04%) events.
The median time from first dose to onset of the event was 1.6 months (range: 14 days to 33.6 months), and the median duration of the event was 5.1 months (range: 4.0 days to 26.4+ months). + denotes a censored observation. Tislelizumab was permanently discontinued in 7 (0.3%) patients and tislelizumab treatment was interrupted in 8 (0.3%) patients.
Ten (66.7%) of the 15 patients received systemic corticosteroids, with a median initial dose of 75 mg/day (range: 20.0 to 200.0 mg/day) for a median duration of 28.5 days (range: 1.0 day to 2.4+ months). + denotes a censored observation. Nine (60%) of the 15 patients received high-dose corticosteroids (defined as a dose ≥ 40 mg/day of prednisone or equivalent). One (6.7%) of the 15 patients received immunosuppressive treatment. Myocarditis resolved in 9 (60%) of the 15 patients. The median duration for all resolved events was 1.2 months (range: 4 days to 15.6 months).
Infusion related reactions. In patients treated with tislelizumab as monotherapy, infusion related reactions occurred in 113 (4.7%) of 2,390 patients, including Grade 1 (74 patients, 3.1%), Grade 2 (36 patients, 1.5%), Grade 3 (2 patients, 0.1%) and Grade 4 (1 patient, 0.04%) events.
Twenty-three (20.4%) of the 113 patients received treatment with corticosteroids. Tislelizumab was permanently discontinued in 4 (0.2%) of patients and tislelizumab treatment was interrupted in 17 (0.7%) of patients. Cases of anaphylaxis, including anaphylactic reaction and anaphylactic shock, have been reported in the post-marketing setting.
Immunogenicity. Overall, a 20.8% incidence (839 of 4035 anti-drug antibody (ADA) evaluable patients) of treatment-emergent ADA was observed following tislelizumab administration across all dose levels, including 0.5 to 10 mg/kg once every 2 weeks, 2 to 5 mg/kg once every 3 weeks, and 200 mg once every 3 weeks (including 200 mg once every 3 weeks in neoadjuvant phase followed by 400 mg once every 6 weeks in adjuvant phase). Neutralising antibodies were detected in 45 of these 4035 patients (1.1%).
Of 3,674 ADA-evaluable patients treated at the recommended dose of 200 mg once every 3 weeks with tislelizumab as monotherapy or in combination with chemotherapies, 776 (21.1%) patients tested positive for treatment-emergent ADAs. The incidence of treatment-emergent ADA was 16.8% (307 of 1822 patients) for monotherapy and 25.3% (469 of 1852 patients) for combination therapy. Neutralising antibodies (NAbs) were detected in 45 (1.2%) patients. The incidence of neutralising antibodies was 1.3% (23 of 1822 patients) for monotherapy and 1.2% (22 of 1852 patients) for combination therapy. Population pharmacokinetic analysis showed that ADA status was a statistically significant covariate on clearance; however, the presence of treatment-emergent ADA against tislelizumab appears to have no clinically relevant impact on pharmacokinetics, efficacy or safety.
Elderly.
No overall differences in safety were observed with tislelizumab monotherapy between patients aged < 65 years and patients aged between 65 and 74 years. Data for patients aged ≥ 75 years are too limited to draw conclusions on this population.
Immune checkpoint inhibitor class effects.
There have been cases of the following adverse reactions reported during treatment with other immune checkpoint inhibitors which might also occur during treatment with tislelizumab: pancreatic exocrine insufficiency, aplastic anaemia.
Reporting of suspected adverse reactions.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no information on overdose with tislelizumab. In case of overdose, patients should be closely monitored for signs or symptoms of adverse drug reactions, and appropriate symptomatic treatment instituted immediately.
For information on the management of overdose, contact the Poisons Information Centre on 131 126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: antineoplastic agents, monoclonal antibodies. ATC code: L01FF09.
Mechanism of action.
Binding of the PD-1 ligands PD-L1 and PD-L2, to the PD-1 receptor found on T-cells, inhibits T-cell proliferation and cytokine production. Upregulation of PD-1 ligands occurs in some tumours and signaling through this pathway can contribute to inhibition of active T-cell immune surveillance of tumours.
Tislelizumab is a humanized immunoglobulin G4 (IgG4) variant monoclonal antibody against PD-1, binding to the extracellular domain of human PD-1 with high specificity and affinity (KD = 0.15 nanoM). It competitively blocks the binding of both PD-L1 and PD-L2, inhibiting PD-1-mediated negative signaling and enhancing the functional activity in T-cells in in vitro cell-based assays. In in vitro studies, tislelizumab did not bind to Fc gamma receptors (FcγRs) and C1q and therefore did not induce antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC); it is not expected to induce antibody-dependent cellular phagocytosis (ADCP). Tislelizumab demonstrated decreased tumour growth in several human cancer allogeneic xenograft models and a human PD-1 transgenic mouse model.
Clinical trials.
Oesophageal squamous cell carcinoma (OSCC). First-line treatment of unresectable, locally advanced, recurrent, locally advanced or metastatic OSCC in combination with chemotherapy.
BGB-A-317-306 is a randomised, double-blind placebo-controlled, global phase III study to compare the efficacy of tislelizumab in combination with platinum-based chemotherapy versus placebo in combination with platinum-based chemotherapy in patients with unresectable, locally advanced recurrent or metastatic OSCC.
The study enrolled patients who were not amenable to chemoradiation or surgery with curative intent. Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, the archival/fresh tumour tissue specimens taken were retrospectively tested for PD-L1 expression status. PD-L1 expression was evaluated using TAP (tumour area positivity) score, defined as the total percentage of the tumour area (tumour and any desmoplastic stroma) covered by tumour cells with PD-L1 membrane staining at any intensity and tumour-associated immune cells with PD-L1 staining at any intensity, as visually estimated using the VENTANA PD-L1 (SP263) assay.
Patients who had received prior systemic therapy for advanced or metastatic disease were excluded. A treatment-free interval of at least 6 months was required if the patient had received prior neoadjuvant/adjuvant therapy with platinum-based chemotherapy.
The study excluded patients who had evidence of fistula or complete oesophageal obstruction not amenable to treatment.
Randomisation was stratified by geographical region (Asia [excluding Japan] versus Japan versus rest of world [ROW]), prior definitive therapy (yes versus no) and investigator choice of chemotherapy (ICC; platinum with fluoropyrimidine or platinum with paclitaxel).
Patients were randomised (1:1) to receive either tislelizumab 200 mg or placebo every 3 weeks in combination with investigator's choice of chemotherapy (ICC) on a 21-day cycle. The chemotherapy doublet regimen consisted of:
platinum (cisplatin [60 to 80 mg/m2 IV on day 1] or oxaliplatin [130 mg/m2 IV on day 1]) and a fluoropyrimidine (5-FU [750 to 800 mg/m2 IV on days 1 to 5] or capecitabine [1000 mg/m2 orally twice daily on days 1 to 14]); or
platinum (cisplatin [60 to 80 mg/m2 IV on day 1 or 2] or oxaliplatin [130 mg/m2 IV on day 1 or 2]) and paclitaxel (175 mg/m2 IV on day 1).
Patients were treated with tislelizumab in combination with chemotherapy or placebo in combination with chemotherapy until disease progression, as assessed by the investigator per RECIST version 1.1 or unacceptable toxicity. After 24 months of treatment, study therapy could be continued beyond two years if the investigator considered this to be in the best interest of the patient based on an assessment of clinical benefit and potential risks.
The tumour assessments were conducted every 6 weeks for the first 48 weeks, and every 9 weeks thereafter.
The primary efficacy endpoint was overall survival (OS) in the intent-to-treat (ITT) population. Secondary efficacy endpoints were progression-free survival (PFS), objective response rate (ORR) and duration of response (DoR) as assessed by the investigator per RECIST v1.1, OS in the PD-L1 positive (PD-L1 score ≥ 10%) subgroup and health-related quality of life (HRQoL).
A total of 649 patients were randomised to receive tislelizumab in combination with chemotherapy (n = 326) or placebo in combination with chemotherapy (n = 323). Of the 649 patients, 293 (45.1%) patients were randomised to receive platinum + fluoropyrimidine, 481 (74.1%) patients had PD-L1 score ≥ 1%, 61 (9.4%) patients had PD-L1 score < 1% and 107 (16.5%) patients had PD-L1 status unknown.
In patients with a PD-L1 score ≥ 1%, the baseline characteristics were: median age 64 years (range: 38 to 84), 47.2% age 65 years or older; 86.3% male; 22.9% White and 75.9% Asian. 86.9% had metastatic disease at study entry and 13.1% had locally advanced disease. All patients had histological confirmation of squamous cell carcinoma. Baseline ECOG performance status was 0 (33.9%) or 1 (66.1%).
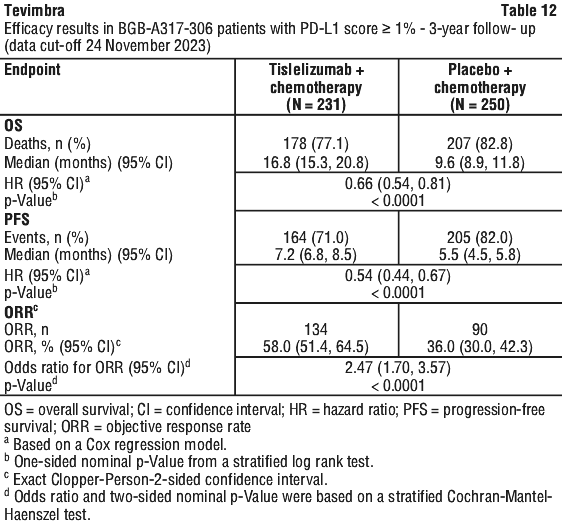
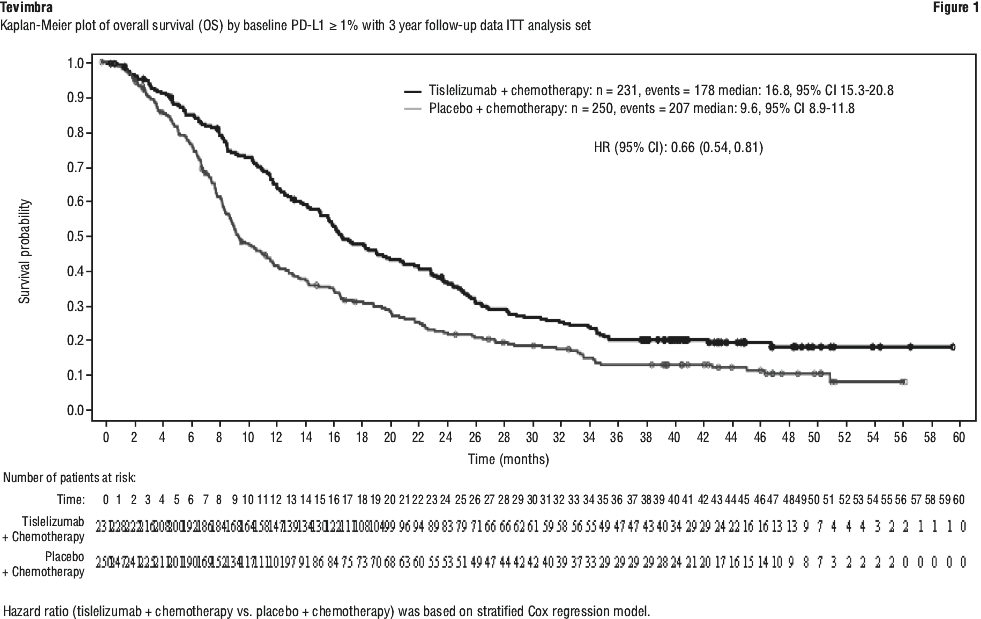
BGB-A317-306 showed a statistically significant improvement in OS for patients randomised to tislelizumab in combination with chemotherapy as compared to placebo in combination with chemotherapy. As of the interim analysis (data cut-off (DCO): 28 February 2022), in patients with a PD-L1 score ≥ 1%, the OS HR was 0.64 (95% CI: 0.51, 0.80, 1-sided p-value of < 0.0001), with a median OS of 16.8 months (95% CI: 15.3, 20.8) for the tislelizumab with chemotherapy arm vs. 9.6 months (95% CI: 8.9, 11.8) for the placebo with chemotherapy arm. The median follow-up times by reverse Kaplan-Meier methodology were 22.8 months in the tislelizumab in combination with chemotherapy arm and 23.3 months in the placebo in combination with chemotherapy arm.
An updated analysis (up to 3-year follow-up; DCO: 24 November 2023) with additional 20.8 months follow-up after the interim analysis showed consistent efficacy results with the interim analysis. The median follow-up times by reverse Kaplan-Meier methodology were 44.2 months in the tislelizumab in combination with chemotherapy arm and 43.8 months in the placebo in combination with chemotherapy arm.
Efficacy results for patients with PD-L1 score ≥ 1%, at 3-year follow-up, are shown in Table 12 and Figure 1.
 In patients with a PD-L1 score ≥ 1% to < 5% (n = 123) (DCO: 24 November 2023) the OS HR was 0.80 (95% CI: 0.52, 1.21) with a median OS of 13.0 months (95% CI: 10.8, 18.3) for the tislelizumab with chemotherapy arm vs 9.6 months (95% CI: 7.9, 13.7) for the placebo with chemotherapy arm. The PFS HR was 0.80 (95% CI: 0.51, 1.24) with a median PFS of 6.5 months (95% CI: 4.7, 7.3) for the tislelizumab with chemotherapy arm vs 5.6 months (95% CI: 4.2, 6.9) for the placebo with chemotherapy arm. The ORR was 40.7% (95% CI: 28.1, 54.3) for the tislelizumab with chemotherapy arm vs 35.9% (95% CI: 35.9 (24.3, 48.9) for the placebo with chemotherapy arm.
In patients with a PD-L1 score ≥ 1% to < 5% (n = 123) (DCO: 24 November 2023) the OS HR was 0.80 (95% CI: 0.52, 1.21) with a median OS of 13.0 months (95% CI: 10.8, 18.3) for the tislelizumab with chemotherapy arm vs 9.6 months (95% CI: 7.9, 13.7) for the placebo with chemotherapy arm. The PFS HR was 0.80 (95% CI: 0.51, 1.24) with a median PFS of 6.5 months (95% CI: 4.7, 7.3) for the tislelizumab with chemotherapy arm vs 5.6 months (95% CI: 4.2, 6.9) for the placebo with chemotherapy arm. The ORR was 40.7% (95% CI: 28.1, 54.3) for the tislelizumab with chemotherapy arm vs 35.9% (95% CI: 35.9 (24.3, 48.9) for the placebo with chemotherapy arm.
In patients with a PD-L1 score ≥ 5% to < 10% (n = 135) (DCO: 24 November 2023) the OS HR was 0.48 (95% CI: 0.31, 0.75) with a median OS of 23.1 months (95% CI: 16.4, 28.3) for the tislelizumab with chemotherapy arm vs 9.8 months (95% CI: 8.0, 13.0) for the placebo with chemotherapy arm. The PFS HR was 0.51 (95% CI: 0.32, 0.82) with a median PFS of 7.2 months (95% CI: 5.7, 15.3) for the tislelizumab with chemotherapy arm vs 4.6 months (95% CI: 4.1, 6.8) for the placebo with chemotherapy arm. The ORR HR was 62.5% (95% CI: 48.5, 75.1) for the tislelizumab with chemotherapy arm vs 36.7% (95% CI: 26.1, 48.3) for the placebo with chemotherapy arm.
Given the limited samples size of these subgroups results should be interpreted with caution.
Patient reported outcomes/quality of life.
Patient-reported outcomes (PROs) were collected using EORTC QLQ-C30, EORTC QLQ-OES18 and EQ-5D-5L questionnaires. Adjusted completion rate from baseline through Cycle 6 and Cycle 8 was ≥ 93% and ≥ 91%, respectively, for each questionnaire in both treatment arms.
No significant differences between treatment arms were observed in the predefined PRO endpoints from baseline to the key clinical Cycle 6. The global health status/QoL and disease-specific symptoms (dysphagia, eating difficulties and reflux) did not worsen with the addition of tislelizumab to chemotherapy treatment, compared with those receiving placebo plus chemotherapy.
Risk of experiencing deterioration for GHS/QoL (HR 0.98 [95% CI 0.74, 1.29]), physical functioning (0.79 [0.60, 1.04]), dysphagia 0.92 (0.70, 1.20), eating 1.00 (0.72, 1.39) and reflux 1.15 (0.83, 1.60) subscales was similar between the treatment groups.
Second-line treatment of unresectable, recurrent, locally advanced or metastatic OSCC.
RATIONALE-302 was a randomised, controlled, open-label, global phase III study to compare the efficacy of tislelizumab versus chemotherapy in patients with unresectable, recurrent, locally advanced or metastatic OSCC who progressed on or after prior systemic treatment. Patients were enrolled regardless of their tumour PD-L1 expression level. Where available, the archival/fresh tumour tissue specimens taken were retrospectively tested for PD-L1 expression status. Patients with inactive or asymptomatic carrier chronic or active HBV status and patients with detectable HCV receiving antivirals at screening were also enrolled in the study.
The study excluded patients with active brain or leptomeningeal tumour invasion into organs located adjacent to the oesophageal disease site (e.g. aorta or respiratory tract), those who had received a prior immune checkpoint inhibitor, active autoimmune disease or history of autoimmune diseases, or any condition requiring systemic treatment with either corticosteroids or other immunosuppressive treatments. PD-L1 expression was evaluated at a central laboratory using the VENTANA PD-L1 (SP263) Assay that identified PD-L1 staining on both tumour and tumour-associated immune cells.
Patients were randomized (1:1) to receive tislelizumab 200 mg every 3 weeks or investigator's choice of chemotherapy (ICC), selected from the following, all given intravenously:
paclitaxel 135 to 175 mg/m2 on day 1, given every 3 weeks (also at doses of 80 to 100 mg/m2 on a weekly schedule according to local and/or country-specific guidelines for standard of care, also administered as 100 mg/m2 on days 1, 8, 15, 22, 29, and 36, followed by 1 week of rest in Japan);
docetaxel 75 mg/m2 on day 1, given every 3 weeks (70 mg/m2 on Day 1, given every 21 days in Japan); or
irinotecan 125 mg/m2 on days 1 and 8, given every 3 weeks.
Crossover between the tislelizumab arm and ICC arm was not permitted. In the ICC arm, switching between the different chemotherapy options was not permitted.
Randomization was stratified by geographic region (Asia [excluding Japan] versus Japan versus USA/EU), ECOG PS score (0 versus 1), and ICC option (paclitaxel versus docetaxel versus irinotecan). The choice of ICC was determined by the investigator before randomization.
Patients were treated with tislelizumab or one of the ICC until disease progression or unacceptable toxicity.
The tumour assessments were conducted every 6 weeks for the first 6 months, and every 9 weeks thereafter. Treatment beyond initial investigator-assessed progression was permitted in patients receiving tislelizumab with no rapid progression, investigator-assessed benefit, tolerance to treatment, stable performance status, and for whom treatment beyond progression would not delay an imminent intervention to prevent serious complications associated with disease progression (e.g. brain metastasis).
The primary efficacy outcome measure was overall survival (OS) in the intent-to-treat (ITT) population. The key secondary efficacy outcome measure was OS in PD-L1 Positive Analysis Set (PD-L1 score ≥ 10%).
Additional secondary efficacy endpoints included objective response rate (ORR), progression-free survival (PFS) and duration of response (DoR), as assessed by the investigator per RECIST v 1.1 and health related quality of life.
A total of 512 patients were enrolled and randomized to tislelizumab (n = 256) or ICC (n = 256: paclitaxel (n = 85), docetaxel (n = 53), or irinotecan (n = 118)). Of the 512 patients, 142 (27.7%) had PD-L1 score ≥ 10%, 222 (43.4%) had PD-L1 score < 10%, and 148 (28.9%) had unknown baseline PD-L1 status.
The baseline characteristics for the 512 patients were: median age of 62 years (range: 35 to 86 years), 37.9% with 65 years of age or older; 84% male; 19% White and 80% Asian; 25% with an eastern cooperative oncology group performance status (ECOG PS) of 0 and 75% with an ECOG PS of 1. 95% of the study population had metastatic disease at study entry. All patients had received at least one prior anti-cancer chemotherapy, which was a platinum-based combination chemotherapy for 97% of patients.
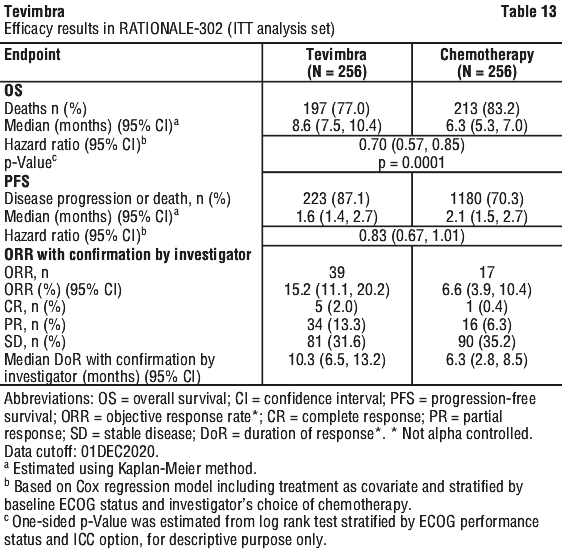
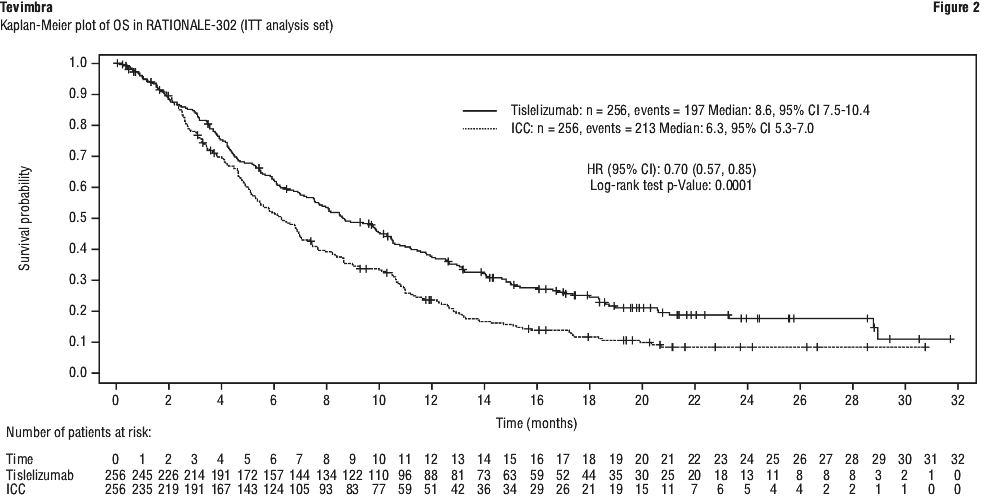
RATIONALE-302 demonstrated a statistically significant improvement in OS for patients randomized to tislelizumab arm as compared with the ICC arm. The median follow-up times of OS by reverse Kaplan-Meier methodology were 20.8 months in the tislelizumab arm and 21.1 months in the ICC arm. Efficacy results are shown in Table 13 and Figure 2.
PD-L1 subgroups.
Of the 512 patients, 142 (27.7%) had PD-L1 positive ESCC, defined as PD-L1 score ≥ 10%. The remaining 222 (43.4%) had PD-L1 negative ESCC defined as PD-L1 score < 10% and 148 (28.9%) had baseline PD-L1 status missing.
In a pre-specified analysis of OS in the PD-L1 positive sub-group (PD-L1 score ≥ 10%), the stratified hazard ratio (HR) for OS was 0.49 (95% CI: 0.33 to 0.74), with a 1-sided stratified log-rank test p-value of 0.0003. The median survival was 10.0 months (95% CI: 8.5 to 15.1 months) and 5.1 months (95% CI: 3.8 to 8.2 months) for the tislelizumab and ICC arms, respectively.
In the PD-L1 negative subgroup (PD-L1 score < 10%), the stratified HR for OS was 0.83 (95% CI: 0.62 to 1.12), with median overall survival of 7.5 months (95% CI: 5.5 to 8.9 months) and 5.8 months (95% CI: 4.8 to 6.9 months) for the Tevimbra arm and ICC arms, respectively.
Non-small cell lung cancer. First-line treatment of metastatic non-squamous NSCLC in combination with pemetrexed and platinum chemotherapy.
The efficacy of tislelizumab was evaluated in RATIONALE-304 (NCT03663205), a multicenter, randomized, open-label, phase 3 study, conducted in China, evaluating the efficacy and safety of tislelizumab combined with chemotherapy in untreated locally advanced non-squamous NSCLC patients who were not candidates for surgical resection or platinum based chemoradiation, or metastatic non-squamous NSCLC patients.
The study excluded patients with active brain or leptomeningeal metastases; with known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (> 10 mg daily of prednisone or equivalent) or other immunosuppressive treatments.
A total of 334 patients were randomized (2:1) to receive tislelizumab 200 mg combined with pemetrexed 500 mg/m2 and carboplatin AUC 5 mg/mL/min or cisplatin 75 mg/m2 (T+PP arm, N = 223), or pemetrexed 500 mg/m2 and carboplatin AUC 5 mg/mL/min or cisplatin 75 mg/m2 (PP arm, N = 111). The choice of platinum (cisplatin or carboplatin) was at the investigator's discretion.
The treatment was administered on a 3-week cycle. After the administration of 4, 5, or 6 cycles of chemotherapy or tislelizumab combined with chemotherapy at the investigator's discretion, patients in arm T+PP received tislelizumab 200 mg combined with pemetrexed 500 mg/m2 on a 3-week cycle until disease progression or unacceptable toxicity. Tislelizumab monotherapy was continued beyond disease progression if the patient was deriving clinical benefit as assessed by the investigator; patients in arm PP received pemetrexed 500 mg/m2 alone until disease progression is confirmed or unacceptable toxicity, and those with disease progression confirmed by IRC given the option to cross over to receive Tevimbra monotherapy on a 3-week cycle.
Randomization was stratified by PD-L1 expression in tumour cells (TC) (< 1% vs 1% to 49% vs ≥ 50%) and disease stage (IIIB vs IV), as classified according to American Joint Committee on Cancer (AJCC), 7th edition of Cancer Staging Manual. PD-L1 expression was evaluated at a central laboratory using the VENTANA PD-L1 (SP263) assay that identified PD-L1 staining on tumour cells. Tumour assessments were conducted every 6 weeks for the first 6 months, then every 9 weeks for the second 6 months, then every 12 weeks.
The baseline characteristics for the study population were: median age of 61 years (range: 25 to 75 years), 29% with 65 years of age or older; 74% male; 100% Asian; 23.4% had an ECOG PS of 0 and 76.6% had an ECOG PS of 1; 18.3% had disease stage IIIb; 26.6% of the patients were unknown status on ALK rearrangement whereas 73.4% with negative ALK rearrangement; 36.2% of the patients were never-smokers; 5.4% with brain metastases, 41.6% PD-L1 TC score < 1%, 24.0% with PD-L1 TC score ≥ 1% and ≤ 49%, 32.9% with PD-L1 TC score ≥ 50%. The characteristics of age, sex, ECOG PS, stage, smoking status, PD-L1 expression and prior anticancer treatments were generally balanced.
The primary efficacy endpoint was progression-free survival (PFS) per RECIST v.1.1 per IRC in the intent to treat (ITT) analysis. The secondary endpoints included overall survival (OS), PFS per investigator, objective response rate (ORR) and duration of response (DoR) per IRC and per investigator.
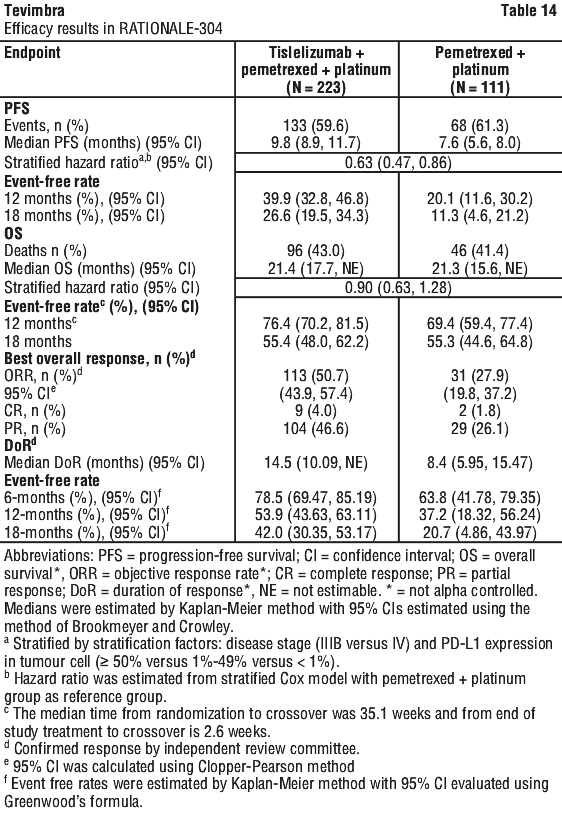
The study met its primary endpoint at the interim analysis (data cut-off date of 23-Jan-2020 and a median duration of study follow-up of 9.0 months) showing a statistically significant improvement in PFS with Tevimbra in combination with PP as compared with PP. The hazard ratio (HR) was 0.65 (95% CI: 0.47, 0.91; p = 0.0054) indicating a 35% reduction in the risk of experiencing disease progression or death, with a median PFS of 9.7 months with Tevimbra in combination with PP and 7.6 months with PP.
The final analysis (data cutoff date of 26-Oct-2020 and a median duration of study follow-up of 16.1 months) were consistent with the results from the interim analysis. Efficacy results for the final analysis are shown in Table 14 and Figure 3.
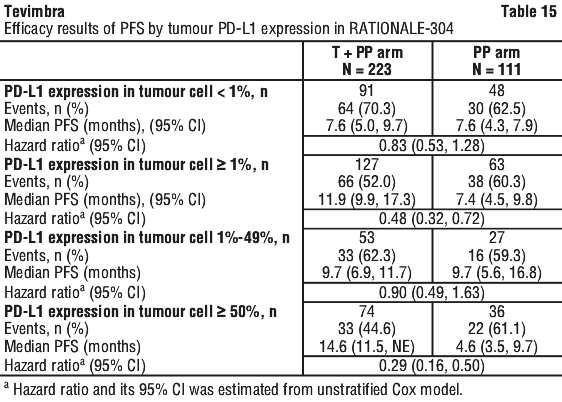
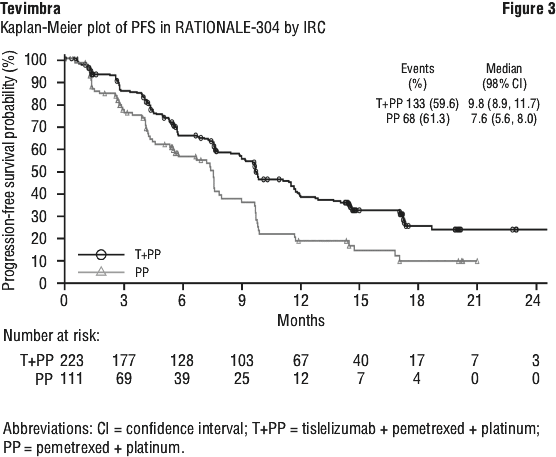 PD-L1 expression in tumor cell < 1%, 1%-49%, ≥ 50% of PFS were pre-specified PFS subgroup analyses, which were not alpha controlled; the results of PFS by tumor PD-L1 expression in the final analysis are shown in Table 15.
PD-L1 expression in tumor cell < 1%, 1%-49%, ≥ 50% of PFS were pre-specified PFS subgroup analyses, which were not alpha controlled; the results of PFS by tumor PD-L1 expression in the final analysis are shown in Table 15.
First-line treatment of locally advanced or metastatic squamous NSCLC in combination with carboplatin and either paclitaxel or nab-paclitaxel chemotherapy.
The efficacy of tislelizumab was evaluated in RATIONALE-307 (NCT03594747), a multicenter, randomized, open-label, phase 3 study, conducted in China, comparing the efficacy and safety of tislelizumab combined with carboplatin and paclitaxel or nab-paclitaxel versus carboplatin and paclitaxel alone as first-line treatment of locally advanced squamous NSCLC patients who were not candidates for surgical resection or platinum based chemoradiation or metastatic NSCLC patients.
The study excluded patients who have active brain or leptomeningeal metastases, known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (> 10 mg daily of prednisone or equivalent) or other immunosuppressive treatments.
A total of 360 patients were randomized (1:1:1) to receive tislelizumab 200 mg combined with paclitaxel 175 mg/m2 and carboplatin AUC 5 mg/mL/min (T+PC arm, N = 120), or tislelizumab 200 mg combined with nab-paclitaxel 100 mg/m2 and carboplatin AUC 5 mg/mL/min (T+nPC arm, N = 119), or paclitaxel 175 mg/m2 and carboplatin AUC 5 mg/mL/min (PC arm, N = 121 arm).
The treatment was administered on a 3-week cycle, until the patient completed administration of 4 to 6 cycles of chemotherapy or tislelizumab combined with chemotherapy at the investigator's discretion. Patients in T+nPC and T+PC arms received tislelizumab until disease progression or unacceptable toxicity; while tislelizumab monotherapy could continue beyond disease progression if the patient was deriving clinical benefit as assessed by the investigator. Patients in the PC arm with disease progression were given the option to cross over to receive tislelizumab monotherapy on a 3-week cycle.
Randomization was stratified by PD-L1 tumour cell (TC) score (< 1% versus 1% to 49% versus ≥ 50%) and tumour staging (IIIB versus IV) as classified according to American Joint Committee on Cancer (AJCC), 7th edition of Cancer Staging Manual. PD-L1 expression was evaluated at a central laboratory using the VENTANA PD-L1 (SP263) assay that identified PD-L1 staining on tumor cells. Tumour assessments were conducted every 6 weeks for the first 6 months, then every 9 weeks for the next 6 months, then every 12 weeks until disease progression.
The baseline characteristics for the study population were: median age 62.0 years (range: 34 to 74 years), 35.3% with 65 years of age or older; 91.7% male; 100% Asian, 23.6% with ECOG PS of 0 and 76.4% with ECOG PS of 1; 33.9% diagnosed with stage IIIB and 66.1% with stage IV at baseline; 16.4% never-smokers; 38.8% with PD-L1 TC score < 1%, 25.3% with PD-L1 TC score ≥ 1% and ≤ 49%, 34.7% with PD-L1 TC score ≥ 50%. The characteristics of age, sex, ECOG PS, stage, smoking status, PD-L1 TC score and prior anticancer treatments were generally balanced.
The primary efficacy endpoint was progression-free survival (PFS) as assessed by IRC per RECIST v1.1 in the intention-to-treat (ITT) analysis which was to be tested sequentially in arms T+PC versus PC and arms T+nPC versus PC. The secondary endpoints included overall survival (OS), objective response rate (ORR) and duration of response (DoR) per IRC and per investigator.
The study met its primary endpoint at the interim analysis (data cut-off date of 06-Dec-2019 and a median duration of study follow-up of 8.4 months), showing statistically significant improvement in PFS with tislelizumab in combination with paclitaxel and carboplatin (T+PC arm) and tislelizumab in combination with nab-paclitaxel and carboplatin (T+nPC arm) compared with paclitaxel and carboplatin alone (PC arm). The stratified HR (T+PC arm versus PC arm) was 0.48 (95% CI: 0.34, 0.69; p < 0.0001), indicating a 52% risk reduction in disease progression or death. The stratified HR was 0.45 (95% CI: 0.32, 0.64; p < 0.0001), a 55% risk reduction in disease progression or death was observed when comparing T+nPC arm with PC arm, with a median PFS of 7.6 months with T+PC arm and 7.6 months with T+nPC arm and 5.4 months with P+C arm.
The final analysis (data cut-off date of 30-Sep-2020 and a median duration of study follow-up of 16.7 months) showed consistent results with the interim analysis. Efficacy results for the final analysis are shown in Table 16, Figure 4 and Figure 5.
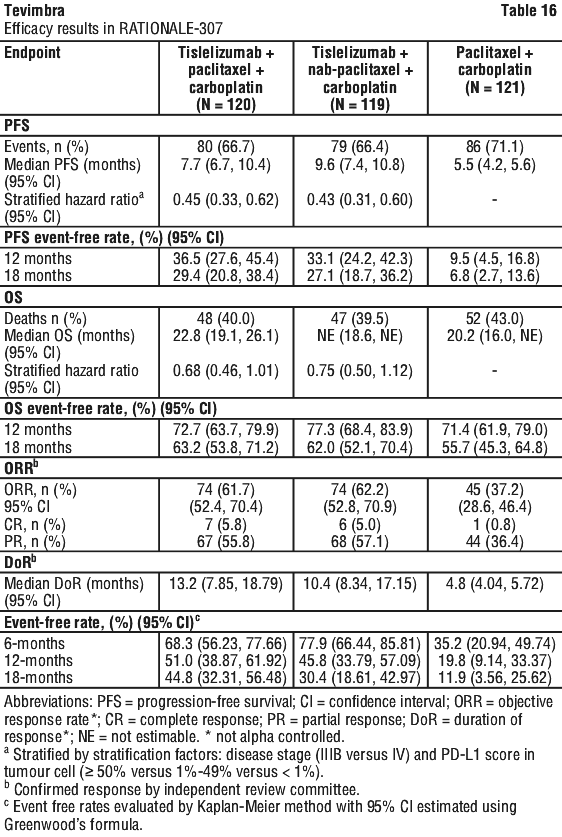
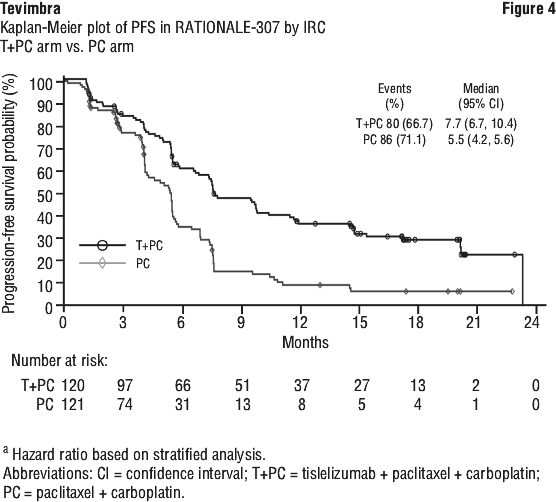
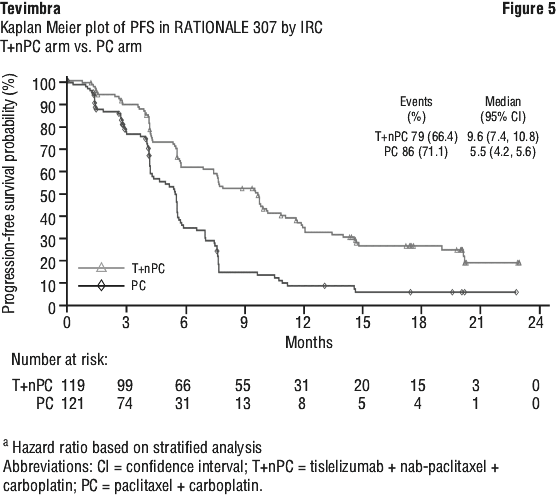 Efficacy results of PFS by tumour PD-L1 expression in pre-specified subgroup analyses for final analysis are shown in Table 17.
Efficacy results of PFS by tumour PD-L1 expression in pre-specified subgroup analyses for final analysis are shown in Table 17.

Previously treated non-small cell lung cancer.
The efficacy of tislelizumab was evaluated in RATIONALE-303 (NCT03358875), a multicenter, randomized, open-label, phase 3 study in patients with locally advanced or metastatic NSCLC (squamous or non-squamous), who had experienced disease progression on or after a prior platinum-based regimen. The study excluded patients with known EGFR mutation or ALK rearrangement, those who had received prior PD-1/PD-L1 antibody treatment, active autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (> 10 mg daily of prednisone or equivalent) or other immunosuppressive treatments.
A total of 805 patients were randomized (2:1) ratio to receive tislelizumab 200 mg intravenously every 3 weeks (N = 535) or docetaxel 75 mg/m2 intravenously every 3 weeks (N = 270). Randomization was stratified by histology (squamous versus non-squamous), lines of therapy (second- versus third-line), and PD-L1 expression in tumour cells (TC) (≥ 25% versus < 25%). Administration of docetaxel and Tevimbra continued until disease progression, as assessed by investigator per RECIST v1.1, or unacceptable toxicity. PD-L1 expression was evaluated at a central laboratory using the VENTANA PD-L1 (SP263) assay that identified PD-L1 staining on tumour cells. Tumour assessments were conducted every 9 weeks for 52 weeks after randomization and continued every 12 weeks thereafter. Survival status was followed every 3 months after discontinuation of the study treatment.
The baseline characteristics for the study population were: median age 61 years (range: 28 to 88 years), 32.4% with 65 years of age or older, 3.2% with 75 years of age or older; 77.3% male; 17.0% White and 79.9% Asian; 20.6% with ECOG PS of 0 and 79.4% with ECOG PS of 1; 85.5% with metastatic disease; 30.3% never smokers; 46.0% with squamous and 54.0% non-squamous histology; 65.8% with wild-type and 34.0% with unknown EGFR status; 46.1% with wild-type and 53.9% with unknown ALK status; 7.1% with brain metastases.
57.0% of the patients had a PD-L1 TC score ≤ 25%, and 42.5% had a PD-L1 TC score ≥ 25%. All patients had received prior therapy with a platinum-doublet regimen, 84.7% patients received one prior therapy, and 15.3% had received two prior therapies. The dual-primary efficacy endpoints were OS in the ITT and PD-L1 TC score ≥ 25% analysis sets. Additional endpoints included investigator assessed PFS, ORR, and DoR.
RATIONALE-303 met both its dual-primary endpoints of OS in the ITT and PD-L1 TC ≥ 25% analysis sets. At the prespecified interim analysis (data cut-off 10-Aug-2020 with a median duration of study follow-up time of 11.7 months), a statistically significant and clinically meaningful improvement in OS was observed in the ITT population. Results favored treatment with tislelizumab, with a 36% relative risk reduction relative to docetaxel (HR = 0.64; 95% CI: 0.53, 0.78; p < 0.0001). Median OS was prolonged by 5.3 months, from 11.9 months for patients receiving docetaxel to 17.2 months for tislelizumab-treated patients.
At the final analysis (data cut-off date of 15-Jul-2021 and a median duration of study follow up of 14.2 months) a statistically significant and clinically meaningful improvement in OS of tislelizumab compared with docetaxel was observed in the PD-L1 TC ≥ 25% analysis set, with a one-sided stratified p < 0.0001. Results favored treatment with Tevimbra, with a 47% relative risk reduction in death relative to docetaxel (stratified HR=0.53; 95% CI: 0.41, 0.70) in the PD-L1 TC ≥ 25% population, with median OS being 19.3 months for the tislelizumab arm and 11.5 months for the docetaxel arm. Efficacy results for the final analysis are shown in Table 18 (ITT analysis set) and Table 19 (PD-L1 TC ≥ 25% analysis set), Figure 6 (ITT analysis set) and Figure 7 (PD-L1 TC ≥ 25% analysis set).
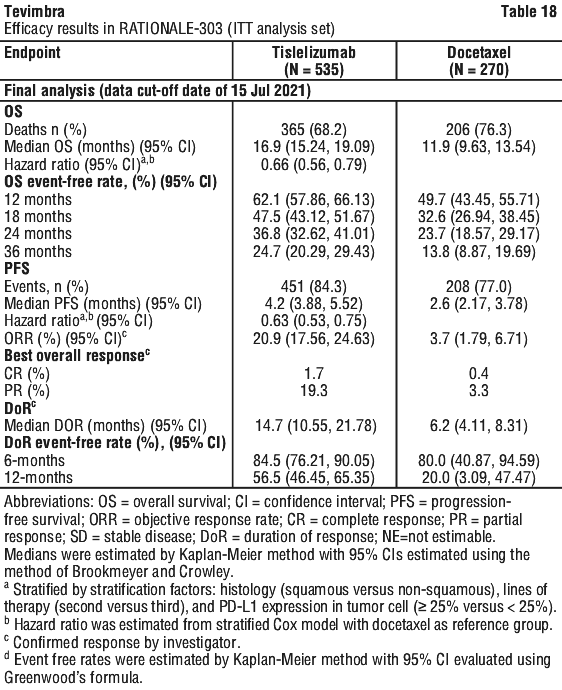
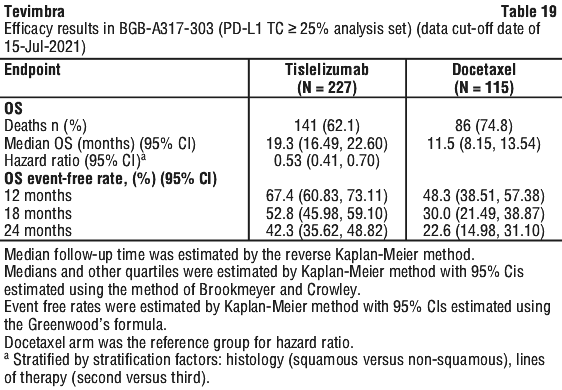
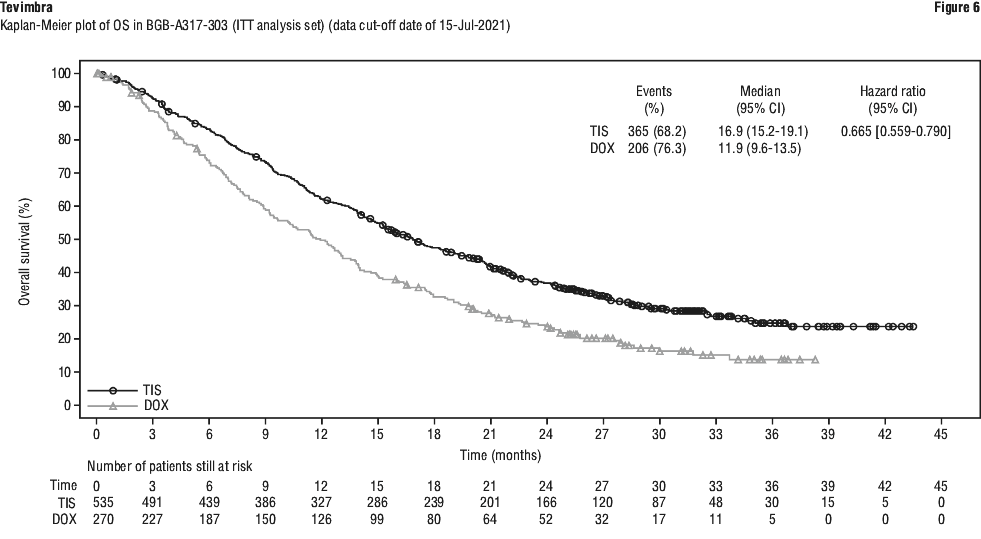
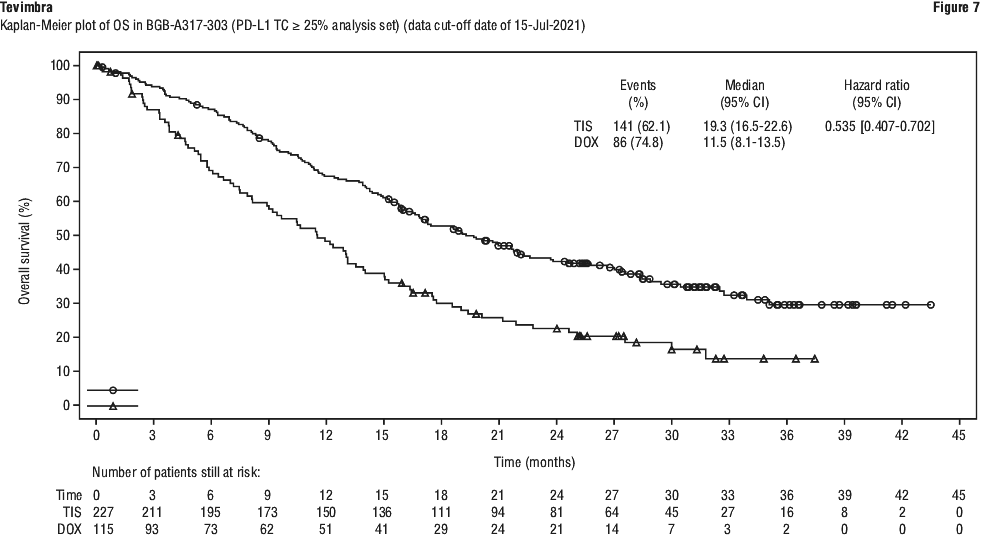 Figure 8 summarises efficacy results of OS by tumour PD-L1 expression in prespecified subgroup analyses at the final analysis.
Figure 8 summarises efficacy results of OS by tumour PD-L1 expression in prespecified subgroup analyses at the final analysis.
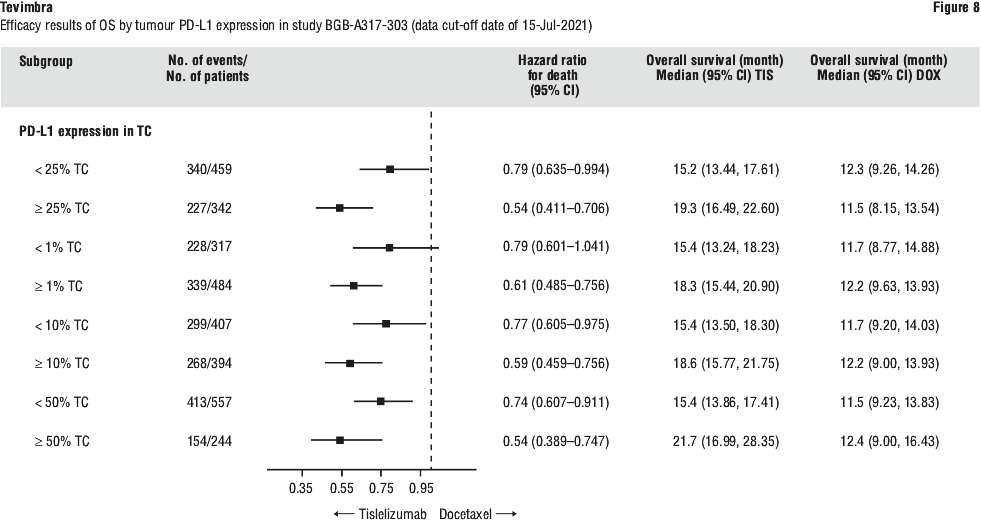 Gastric/gastro-oesophageal junction (G/GOJ) adenocarcinoma.
Gastric/gastro-oesophageal junction (G/GOJ) adenocarcinoma. First-line treatment of patients with HER-2 negative locally advanced unresectable or metastatic G/GOJ adenocarcinoma in combination with platinum and fluoropyrimidine-based chemotherapy.
RATIONALE-305 (NCT03777657) is a randomised, multicentre, placebo-controlled, double-blind trial in patients with previously untreated locally advanced unresectable or metastatic G/GOJ adenocarcinoma.
Patients were enrolled regardless of their tumour PD-L1 expression level, which was evaluated prospectively at a central laboratory using the VENTANA PD-L1(SP263) assay that identified PD-L1 staining on both tumour and tumour-associated immune cells (Tumour Area Positivity (TAP) score).
The study included only patients with histologically confirmed adenocarcinoma. The trial excluded patients who had squamous cell or undifferentiated or other histological type of G/GOJ cancer; patients diagnosed with G/GOJ adenocarcinoma with known human epidermal growth factor receptor 2 (HER2) positive tumours; patients who had active leptomeningeal disease or uncontrolled brain metastasis; patients with active autoimmune disease or history of autoimmune diseases, that may relapse.
Tevimbra plus platinum and fluoropyrimidine based chemotherapy (T+PtF) or placebo plus platinum and fluoropyrimidine based chemotherapy (P+PtF) were administered on a 21-day cycle. Tevimbra (or placebo) was administered until disease progression or unacceptable toxicity.
The administration of chemotherapy agents consisted of:
Oxaliplatin 130 mg/m2 IV on day 1, and capecitabine 1000 mg/m2 orally twice daily for 14 consecutive days, repeated every 3 weeks. Oxaliplatin was administered for up to 6 cycles and capecitabine was administered as maintenance therapy at investigators discretion until disease progression or unacceptable toxicity; or
Cisplatin 80 mg/m2 IV on Day 1, and 5-FU 800 mg/m2/day IV continuous infusion over 24 hours daily on Day 1-5, repeated every 3 weeks. Cisplatin and 5-FU were given for up to 6 cycles.
Cross-over between treatment arms was not allowed.
Randomisation was stratified by region (China (including Taiwan), vs. Japan and South Korea vs. rest of the world (ROW), including US and Europe); PD-L1 expression (PD-L1 TAP score > 5% vs. PD-L1 TAP score < 5%); presence of peritoneal metastasis (yes vs. no) and ICC option (oxaliplatin plus capecitabine vs. cisplatin plus 5-FU).
Additional analyses of efficacy outcome measures were also conducted based on PD-L1 TAP score > 1%.
The primary efficacy outcome measures were OS that was tested hierarchically in the PD-L1 TAP score > 5% population and in the Intent-to-Treat (ITT) population.
Secondary outcome measures included progression-free survival (PFS), objective response rate (ORR), and duration of response (DoR) as assessed by the investigator per RECIST v1.1. Tumour assessments were performed every 6 weeks for the first 48 weeks and thereafter approximately every 9 weeks.
A total of 997 patients were randomized to receive either tislelizumab plus platinum and fluoropyrimidine based chemotherapy (T+PtF arm, N = 501), or placebo plus platinum and fluoropyrimidine based chemotherapy (P+PtF arm, N = 496). Of the 997 patients, 885 (88.8%) had PD-L1 TAP score ≥ 1% (T+PtF arm, N = 432; P+PtF arm, N = 453).
The demographics and baseline characteristics of PD-L1 TAP ≥ 1% population were generally balanced in T+PtF and P+PTF arms. 34.5% patients in T+PtF Arm and 37.1% patients in P+PtF Arm were aged 65 years or older. 65.5% patients in T+PtF arm and 68.0% patients in P+ PtF arm had ECOG performance status score of 1 at baseline. 24.8% patients in T+PtF arm and 25.4% patients in P+PtF arm were enrolled from US/Europe. Most patients had metastatic disease at baseline (98.6% in T+PtF arm and 99.1% in P+PtF arm), while 44.0% patients in T+PtF arm and 43.3% patients in P+PtF arm had peritoneal metastasis.
At final analysis (DCO: 28 February 2023), with a minimum follow-up time of 24.6 months, the trial met the primary endpoint of OS in the ITT analysis set, demonstrating a statistically significant and clinically meaningful improvement for the ITT population. In the PD-L1 TAP ≥ 1% population, the OS HR was 0.78 (95% CI: 0.67, 0.90), with a median OS of 15.0 months (95% CI: 13.3, 16.7) for the T+C arm vs. 12.8 months (95% CI: 12.1, 14.1) for the P+C arm, showing clinical meaningful survival benefit favouring T+C arm.
The median PFS was 6.9 months in T+PtF arm and 5.9 months in P+PtF arm, with HR of 0.78 (95% HR 0.67, 0.91). The ORR was 47.7% in T+PtF arm and 41.1% in P+PtF arm, with odds ratio of 1.31 (95%CI: 1.00, 1.72). For patients with response, the median DoR was 8.6 months T+PtF arm and 7.2 months in P+PtF arm. Analysis of these efficacy endpoints also demonstrated better disease control in T+PtF arm in this population.
Final analysis efficacy results of PD-L1 TAP ≥ 1% population are shown in Table 20, and Figure 9 and 10.
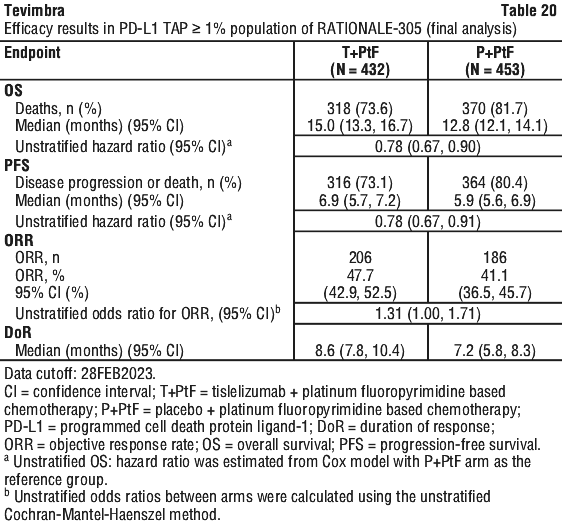
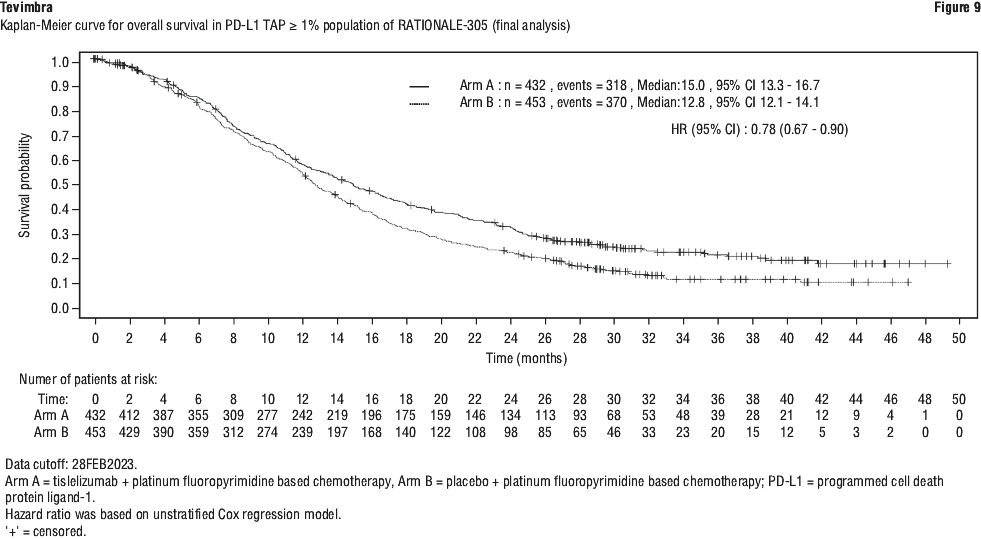
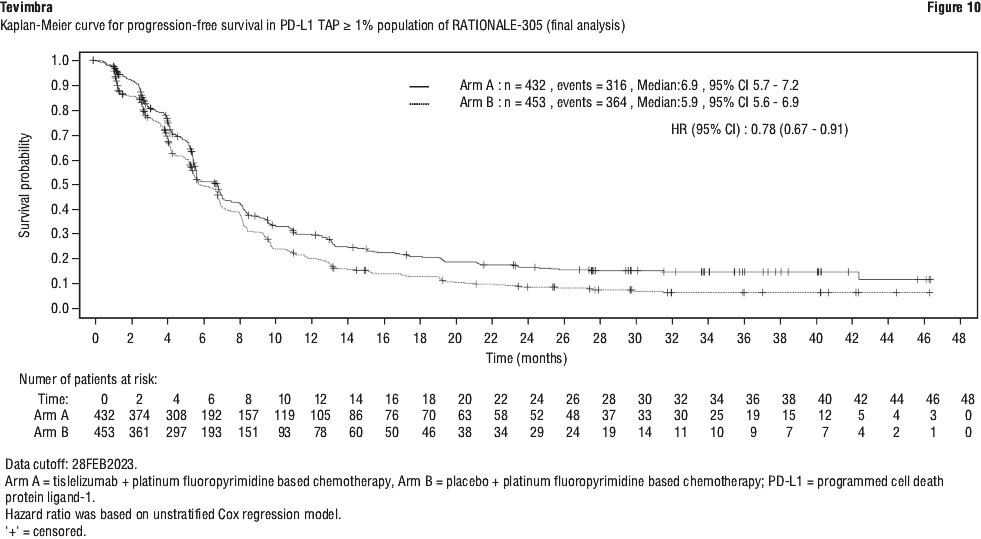 Nasopharyngeal carcinoma (NPC).
Nasopharyngeal carcinoma (NPC). First-line treatment of patients with recurrent or metastatic NPC in combination with gemcitabine and cisplatin.
RATIONALE-309 (NCT03924986) is a randomised, multicentre, double-blind, placebo-controlled Phase 3 study comparing the efficacy and safety of Tevimbra in combination with gemcitabine and cisplatin versus placebo in combination with gemcitabine and cisplatin as first-line treatment in patients with recurrent or metastatic nasopharyngeal carcinoma (NPC).
A total of 263 patients were randomised (1:1) to receive either Tevimbra 200 mg in combination with gemcitabine and cisplatin (N = 131) or placebo in combination with gemcitabine and cisplatin (N = 132).
The study included patients with pathologically confirmed recurrent or metastatic NPC who have not received prior systemic treatment and an ECOG performance status of 0 or 1.
The RATIONALE-309 study excluded patients who received any approved systemic anticancer therapy including hormonal therapy within 28 days before initiation of study treatment, prior therapies targeting PD-1 or PD-L1, or who have active leptomeningeal disease or uncontrolled, untreated brain metastasis.
The baseline characteristics for the study population were: median age of 50 years (range: 23 to 74 years); 91.6% of patients < 65 years old; 78.3% of patients were male; 63.1% with baseline ECOG PS score of 1; 100% Asian (from China, Thailand, and Taiwan); and 95% were never or former smokers. Histological subtypes of NPC included 86.3% non-keratinising, 6.5% keratinising squamous cell carcinoma, and 7.2% did not have their subtype classified. 95.1% of patients had metastatic disease. The baseline demographic and disease characteristics in the ITT analysis set were generally well-balanced between the 2 arms.
Randomisation was stratified by gender and metastatic status (with versus without liver metastases). Patients were randomised (1:1) to receive either Tevimbra 200 mg or placebo plus cisplatin 80 mg/m2 on Day 1 plus gemcitabine 1 g/m2 on Day 1 and Day 8 of each 21-day cycle for 4 to 6 cycles. Tevimbra or placebo was administered until disease progression or unacceptable toxicity. For patients in placebo plus gemcitabine and cisplatin arm, crossover to monotherapy Tevimbra was allowed after disease progression confirmed by the Independent Review Committee (IRC).
The primary endpoint was progression-free survival (PFS) as assessed by the IRC per RECIST v1.1 in the Intention-to-Treat (ITT) analysis set. Secondary endpoints included Overall Survival (OS), Objective Response Rate (ORR) assessed by IRC, and Duration of Response (DOR) assessed by IRC.
At interim analysis (data cut-off date 26-Mar-2021) with a minimum follow-up time of 10.0 months, RATIONALE-309 demonstrated a statistically significant improvement in PFS for patients randomised to the Tevimbra in combination with gemcitabine and cisplatin arm compared with the placebo plus gemcitabine and cisplatin arm.
Interim analysis efficacy results are shown in Table 21 and Figure 11.
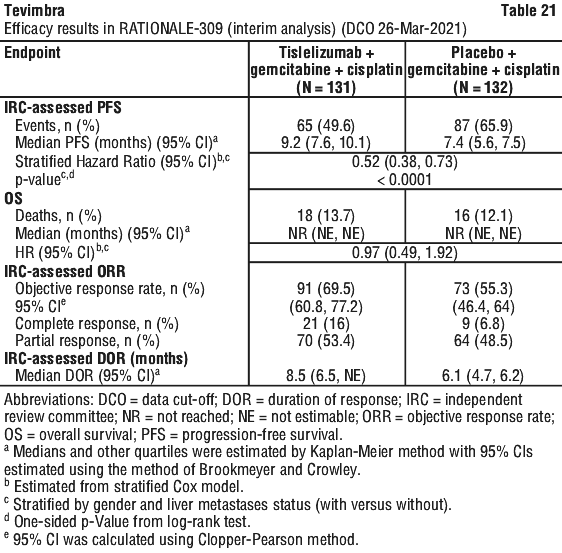
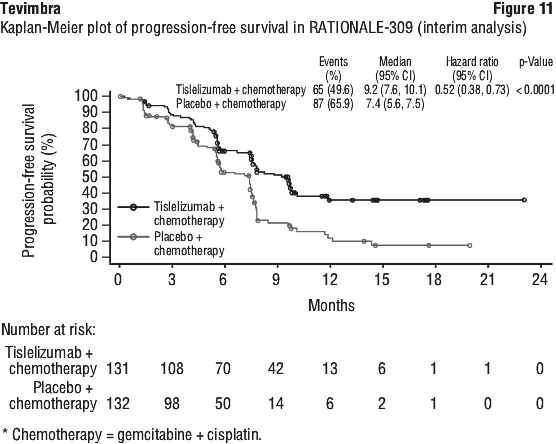
5.2 Pharmacokinetic Properties
The pharmacokinetics (PK) of tislelizumab was assessed for tislelizumab both as monotherapy and in combination with chemotherapy.
The PK of tislelizumab was characterised using population PK analysis with concentration data from 2,596 patients with advanced malignancies who received tislelizumab doses of 0.5 to 10 mg/kg every 2 weeks, 2.0 and 5.0 mg/kg every 3 weeks, and 200 mg every 3 weeks.
Tislelizumab exhibited linear PK in the dose range tested (0.5 mg/kg to 10 mg/kg, including 200 mg flat dose). The time to reach 90% steady-state level is approximately 84 days (12 weeks) after 200 mg once every 3 weeks (Q3W) doses, and the steady-state accumulation ratio of tislelizumab PK exposure is approximately 2-fold.
Absorption.
Tislelizumab is administered via the intravenous route and therefore is immediately and completely bioavailable.
Distribution.
A population pharmacokinetic analysis indicates that the steady state volume of distribution is 6.42 L, which is typical of monoclonal antibodies with limited distribution.
Metabolism.
Tislelizumab is expected to be degraded into small peptides and amino acids via catabolic pathways.
Excretion.
Based on population PK analysis, the clearance of tislelizumab was 0.153 L/day with an inter-individual variability of 26.3% and the geometrical mean terminal half-life was approximately 23.8 days with a coefficient variation (CV) of 31%. Time-varying clearance was not observed in tislelizumab PK.
Linearity/non-linearity.
At the dosing regimens of 0.5 mg/kg to 10 mg/kg once every 2 or 3 weeks (including 200 mg once every 3 weeks), PK of tislelizumab were observed to be linear and dose proportional, suggesting saturation of the target-mediated pathway.
Special populations.
The effects of various covariates on the PK of tislelizumab were assessed in population PK analyses. The following factors had no clinically relevant effect on the exposure of tislelizumab: age (range 18 to 90 years), weight (range 32 to 130 kg), gender, race (White, Asian, and other), mild to moderate renal impairment (creatinine clearance (ClCr) ≥ 30 mL/min), mild to moderate hepatic impairment (total bilirubin ≤ 3 times ULN and any AST), and tumour burden.
Elderly patients.
Of 2,596 patients who received tislelizumab as monotherapy or combination therapies, 1,750 patients (67.4%) were < 65 years and 846 (32.6%) patients were ≥ 65 years of age, 737 (28.4%) patients between 65 and 75 years, and 109 (4.2%) patients > 75 years).
Based on population PK and exposure-response analysis, no clinically relevant differences in PK or safety or efficacy of tislelizumab were observed between patient's aged < 65 years, patients aged between 65 and 75 years and patients aged > 75 years (see Section 4.2 Dose and Method of Administration).
Patients with renal impairment.
No dedicated studies of tislelizumab have been conducted in patients with renal impairment. In the population PK analyses of tislelizumab, no clinically relevant differences in the clearance of tislelizumab were found between patients with mild renal impairment (ClCr) 60 to 89 mL/min, n = 1,046) or moderate renal impairment (ClCr 30 to 59 mL/min, n = 320) and patients with normal renal function (ClCr ≥ 90 mL/min, n = 1223). Mild and moderate renal impairment had no effect on the exposure of tislelizumab (see Section 4.2 Dose and Method of Administration). Based on the limited number of patients with severe renal impairment (n = 5), the effect of severe renal impairment on the pharmacokinetics of tislelizumab is not conclusive.
Patients with hepatic impairment.
No dedicated studies of tislelizumab have been conducted in patients with hepatic impairment. In the population PK analyses of tislelizumab, no clinically important differences in the clearance of tislelizumab were found between patients with mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1.0 to 1.5 x ULN and any AST, n = 396), moderate hepatic impairment (bilirubin > 1.5 to 3 x ULN and any AST, n = 12), compared to patients with normal hepatic function (bilirubin ≤ ULN and AST ≤ ULN, n = 2,182) (see Section 4.2 Dose and Method of Administration). Based on the limited number of patients with severe hepatic impairment (n = 2), the effect of severe hepatic impairment on the pharmacokinetics of tislelizumab is unknown.
Hepatic impairment was defined by the National Cancer Institute-Organ Dysfunction Working Group (NCI-ODWG) criteria of hepatic dysfunction.
5.3 Preclinical Safety Data
Genotoxicity and carcinogenicity.
No studies have been performed to assess the potential of tislelizumab for carcinogenicity or genotoxicity.6 Pharmaceutical Particulars
6.1 List of Excipients
Sodium citrate dihydrate, citric acid monohydrate, histidine hydrochloride monohydrate, histidine, trehalose dihydrate, polysorbate-20, and water for injection (WFI).
6.2 Incompatibilities
This product must not be mixed with products except sodium chloride, which is used to prepare diluted solution.
6.3 Shelf Life
Unopened vial.
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
After opening.
Once opened, the medicinal product should be diluted and infused immediately (see Section 4.2 Dose and Method of Administration, Instructions for use and handling for instructions on dilution of the medicinal product before administration).
After preparation of infusion.
Chemical and physical in-use stability has been demonstrated for 10 days (240 hours at 2°C to 8°C). The 10 days includes storage of the diluted solution under refrigeration (2°C to 8°C), time required for returning to room temperature (25°C or below) and time to complete the infusion within 4 hours.
6.4 Special Precautions for Storage
Store in a refrigerator at 2 to 8°C. Do not freeze. Store in the original carton box to protect from light.
For storage conditions after dilution of the medicinal product, see Section 6.3 Shelf Life.
6.5 Nature and Contents of Container
10 mL of Tevimbra concentrate is provided in a 20 mL clear Type 1 glass vial, with a grey chlorobutyl stopper with FluroTec coating and seal cap with a flip-off button.
Each carton contains one vial.
6.6 Special Precautions for Disposal
Disposal.
Tevimbra is for single use in one patient only. Discard any residue.6.7 Physicochemical Properties
Chemical structure.
Tevimbra (tislelizumab) is a humanized monoclonal antibody that binds with high affinity to the human programmed cell death protein 1 (PD-1). Tislelizumab is an immunoglobulin subclass 4 (IgG4) variant produced by recombinant DNA technology in Chinese hamster ovary cells, with an approximate molecular weight of 144 kDa.
CAS number.
1858168-59-8.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes
