1 Name of Medicine
Ivosidenib.
2 Qualitative and Quantitative Composition
Each film-coated tablet contains 250 mg of ivosidenib.
Excipient with known effect.
Contains lactose. For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Blue, oval shaped, film-coated tablets approximately 18 mm in length, debossed with 'IVO' on one side and '250' on the other side.
4.1 Therapeutic Indications
Cholangiocarcinoma.
Tibsovo is indicated for the treatment of adult patients with locally advanced or metastatic cholangiocarcinoma with an isocitrate dehydrogenase-1 (IDH1) R132 mutation after at least one prior line of systemic therapy.
Acute myeloid leukaemia.
Tibsovo is indicated for the treatment of acute myeloid leukaemia (AML) that carries an IDH1 R132 mutation:
as monotherapy, or in combination with azacitidine, in newly diagnosed patients who are not eligible to receive intensive induction chemotherapy; or
as monotherapy in patients whose AML is relapsed and/or refractory to prior therapy.4.2 Dose and Method of Administration
Treatment should be initiated by a physician experienced in the use of anti-cancer therapies. Before taking Tibsovo, patients must have confirmation of an IDH1 mutation using an appropriate diagnostic test, and an electrocardiogram (ECG) to assess heart rate corrected QT (QTc) interval. Patients with AML without IDH1 mutations at diagnosis should be retested at relapse because a mutation in IDH1 may emerge during treatment or at relapse.
Dose.
The recommended dose of Tibsovo is 500 mg orally once daily until disease progression or unacceptable toxicity.
When Tibsovo is used in combination with azacitidine to treat patients with newly diagnosed AML, the recommended dose of azacitidine is 75 mg/m2 of body surface area, intravenously or subcutaneously, once daily on Days 1-7 (or on Days 1-5, then on Days 8 and 9) of each 28-day cycle. Refer to the full product information for azacitidine for additional dosing information.
Continue treatment for AML (whether monotherapy or in combination with azacitidine) for a minimum of six months to allow time for clinical response.
Method of administration.
Tibsovo should be taken at about the same time each day, with or without food, but not with a high fat meal (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 5.2 Pharmacokinetic Properties). Do not split, crush or chew the tablets.
Two doses should not be taken within 12 hours. If a dose of Tibsovo is missed or not taken at the usual time, administer the dose as soon as possible within 12 hours after it was missed. Administer the following day's dose at the usual time. If 12 hours or longer have elapsed since a dose was missed, do not administer the dose; wait until the next scheduled dose is due. If a dose of Tibsovo is vomited, do not administer replacement tablets; wait until the next scheduled dose is due.
Monitoring.
QTc interval prolongation.
Perform an ECG at baseline, at least weekly during the first three weeks of therapy and at least monthly thereafter. Monitor electrolytes at baseline and throughout treatment as clinically indicated. Patients at higher risk of QTc interval prolongation, including due to concomitant medications, may require more frequent monitoring. Promptly manage abnormalities (see Table 1 and see Section 4.4 Special Warnings and Precautions for Use).
Differentiation syndrome in AML.
Assess full blood count and blood chemistry prior to initiating treatment, and then as clinically indicated: including at least weekly for the first month, at least fortnightly for the second month, and at least monthly thereafter. Promptly manage abnormalities (see Table 1 and see Section 4.4 Special Warnings and Precautions for Use).
Dose modification for concomitant administration of strong CYP3A4 inhibitors.
If use of strong CYP3A4 inhibitors is unavoidable, reduce the Tibsovo dose to 250 mg once daily. If the strong CYP3A4 inhibitor is discontinued, increase the Tibsovo dose to 500 mg after at least 5 half-lives of the strong CYP3A4 inhibitor (see above and see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Dose modifications for adverse reactions.
Guidelines for management in case of adverse reactions are summarised in Table 1. Also see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 4.8 Adverse Effects (Undesirable Effects).

Special populations.
Renal impairment.
No dose adjustment is required in patients with mild (eGFR ≥ 60 to < 90 mL/min/1.73 m2) or moderate (eGFR ≥ 30 to < 60 mL/min/1.73 m2) renal impairment. A recommended dose has not been determined for patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2). See Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties.
Hepatic impairment.
No dose adjustment is required in patients with mild or moderate hepatic impairment (Child-Pugh class A or B). No studies have been conducted in patients with severe hepatic impairment (Child-Pugh class C) and a recommended dose has not been determined in this population. See Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties.
Elderly population.
No dose adjustment is required in elderly patients (≥ 65 years old). See Section 4.8 Adverse Effects (Undesirable Effects); Section 5.2 Pharmacokinetic Properties.
Paediatric population.
No data are available. See Section 4.4 Special Warnings and Precautions for Use.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Differentiation syndrome in patients with acute myeloid leukaemia (AML).
Tibsovo treatment can cause differentiation syndrome in patients with AML (see Section 4.8 Adverse Effects (Undesirable Effects)), in keeping with its mechanism of action. Differentiation syndrome is associated with rapid proliferation and differentiation of myeloid cells and may be life-threatening or fatal.
Symptoms of differentiation syndrome in patients treated with Tibsovo in pivotal studies included noninfectious leukocytosis, peripheral oedema, pyrexia, dyspnoea, pleural effusion, hypotension, hypoxia, pulmonary oedema, pneumonitis, pericardial effusion, rash, fluid overload, tumour lysis syndrome and creatinine increased. The timing of onset of differentiation syndrome follows that of AML response to treatment: it can occur within a day or after many months of treatment.
If differentiation syndrome is suspected, commence systemic corticosteroids (dexamethasone 10 mg IV every 12 hours or an equivalent dose of an alternative oral or IV corticosteroid) and initiate haemodynamic monitoring. If noninfectious leukocytosis is also observed, initiate treatment with hydroxycarbamide or leukapheresis as clinically indicated.
Continue corticosteroids for a minimum of three days, and taper corticosteroids and hydroxycarbamide only after resolution of symptoms. Symptoms of differentiation syndrome may recur with premature discontinuation of corticosteroid and/or hydroxycarbamide treatment.
If severe (Grade 3 or higher) signs/symptoms persist for more than 48 hours after the initiation of systemic corticosteroids, interrupt Tibsovo until signs/symptoms are no longer severe (see Section 4.2 Dose and Method of Administration).
QTc interval prolongation.
Tibsovo causes prolongation of the QTc interval, and ventricular arrhythmias have been reported following treatment with Tibsovo in patients with haematological malignancies (see Section 5.1 Pharmacodynamic Properties; Section 4.8 Adverse Effects (Undesirable Effects)). Perform an ECG prior to treatment initiation, at least weekly during the first 3 weeks of therapy and at least monthly thereafter and monitor electrolytes. Manage any abnormalities promptly (see Section 4.2 Dose and Method of Administration).
Avoid concomitant administration of medicines known to prolong the QTc interval (e.g. antiarrhythmics, fluoroquinolones, 5-HT3 receptor antagonists, triazole antifungals), or moderate or strong CYP3A4 inhibitors, as these may increase the risk of QTc interval prolongation (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). If concomitant use is unavoidable, or for patients with other risk factors (such as congenital long QTc syndrome, congestive heart failure or electrolyte abnormalities), monitor closely, with more frequent ECGs and regular monitoring of electrolytes as required. Adjust dosing if concomitant use of a strong CYP3A4 inhibitor is unavoidable (see Section 4.2 Dose and Method of Administration).
Interrupt Tibsovo for QTc interval over 480 msec, and permanently discontinue Tibsovo in patients with QTc interval prolongation and signs or symptoms of life-threatening arrhythmia (see Section 4.2 Dose and Method of Administration).
Guillain-Barré syndrome.
Guillain-Barré syndrome has occurred uncommonly in patients with haematological malignancies treated with Tibsovo. A causal mechanism is not known, and preclinical studies did not identify the CNS as a target organ for ivosidenib toxicity. No cases of Guillain-Barré syndrome have been reported in patients with solid tumours, though peripheral neuropathy is common (see Section 4.8 Adverse Effects (Undesirable Effects)).
Monitor patients taking Tibsovo for onset of new signs or symptoms of motor and/or sensory neuropathy such as unilateral or bilateral weakness, sensory alterations, paraesthesias, or difficulty breathing. Permanently discontinue Tibsovo in patients who are diagnosed with Guillain-Barré syndrome.
Use in renal impairment.
The safety and efficacy of Tibsovo have not been established in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2), including those requiring dialysis. Use Tibsovo with caution and monitor closely in this population (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Use in hepatic impairment.
The safety and efficacy of Tibsovo have not been established in patients with severe hepatic impairment (Child-Pugh class C). Use Tibsovo with caution and monitor closely in this population (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
No overall differences in effectiveness or safety were observed in patients ≥ 65 years of age (see Section 4.8 Adverse Effects (Undesirable Effects)).
Paediatric use.
The safety and efficacy of Tibsovo in children and adolescents < 18 years old have not been established. No data are available.
Effects on laboratory tests.
See Table 4, Table 6, Table 8 and Table 10 in Section 4.8 Adverse Effects (Undesirable Effects).
Reproductive toxicity.
Tibsovo may cause fetal harm if administered during pregnancy. Verify pregnancy status prior to starting treatment and advise the use of barrier contraception as ivosidenib may decrease systemic concentrations of hormonal contraceptives (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 4.6 Fertility, Pregnancy and Lactation).
Interactions.
Clinically significant interactions are predicted with Tibsovo. Give advice regarding potential for food interactions and review concomitant medications (see QTc interval prolongation and Reproductive toxicity, above; see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 5.2 Pharmacokinetic Properties).4.5 Interactions with Other Medicines and Other Forms of Interactions
Summary of interactions.
Co-administration of Tibsovo with certain medicines and foods is likely to lead to clinically significant interactions. Categories of substances that interact (or may interact) with ivosidenib are summarised in Table 2 though the included examples are not an exhaustive list.

Effect of other medicines on Tibsovo.
Strong CYP3A4 inducers.
Ivosidenib is a CYP3A4 substrate. Physiologically based pharmacokinetic (PBPK) modelling predicted a 33% decrease in ivosidenib steady-state AUC (AUCss) when given at the recommended dose in the presence of co-administered 600 mg rifampin once daily for 15 days. Avoid co-administration of strong CYP3A4 inducers (see Table 2).
Moderate or strong CYP3A4 inhibitors.
Co-administration of a single dose of Tibsovo 250 mg with a strong CYP3A4 inhibitor (200 mg itraconazole daily for 18 days) increased the ivosidenib AUC by 2.7-fold (with no change in Cmax) in healthy volunteers. PBPK modelling predicted an increase in ivosidenib AUCss in the presence of a co-administered strong (ketoconazole: 3.2-fold) or moderate (fluconazole: 1.9-fold) CYP3A4 inhibitor. Avoid co-administration of moderate or strong CYP3A4 inhibitors (see Table 2): consider alternative therapies. If co-administration is unavoidable, treat with caution and monitored closely for QTc interval prolongation (see Section 4.4 Special Warnings and Precautions for Use). If co-administration of a strong CYP3A4 inhibitor is unavoidable, reduce the ivosidenib dose (see Section 4.2 Dose and Method of Administration).
Interactions with transporters.
Ivosidenib is a P-glycoprotein (P-gp) substrate. However, data from study in healthy subjects suggest that the potential for clinically relevant interactions with ivosidenib and P-gp inhibitors is low.
Effect of Tibsovo on other medicines.
Enzyme induction.
Ivosidenib induces CYP3A4 (including its own metabolism), CYP2B6, CYP2C8, CYP2C9 and may induce CYP2C19 and UGT (see Section 5.2 Pharmacokinetic Properties). Therefore, it may decrease systemic exposure to substrates of these enzymes. This is of particular importance for substrates with a narrow therapeutic index or with significant clinical consequence of inefficacy (such as hormonal contraceptives and antifungals: see Table 2). Consider suitable alternatives, recommend barrier contraception (see Section 4.6 Fertility, Pregnancy and Lactation), and if concomitant use can't be avoided, monitor for loss of substrate efficacy.
Interactions with transporters.
Ivosidenib inhibits P-gp and OAT3 and has the potential to induce P-gp. Therefore, it may alter systemic exposure to active substances that are predominantly transported by P-gp and may increase systemic exposure to OAT3 substrates (see Table 2). Consider suitable alternatives, and if concomitant use can't be avoided, monitor for loss of substrate efficacy or P-gp substrate toxicity. Avoid co-administration of dabigatran due to risk of dabigatran toxicity (haemorrhage).
Other interactions.
Medicines known to prolong the QTc interval.
Co-administration of other medicines known to prolong the QTc interval (see Table 2) may increase the risk of QTc interval prolongation. Avoid co-administration of medicines known to prolong the QTc interval (see Table 2): consider alternative therapies. If co-administration is unavoidable, treat with caution and monitored closely for QTc interval prolongation (see Section 4.4 Special Warnings and Precautions for Use).
Food interactions.
Administration of Tibsovo with a high-fat meal should be avoided, as it has a significant effect on the absorption of ivosidenib and leads to increased exposure (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Grapefruit and grapefruit juice moderately inhibit CYP3A4 (see Moderate or strong CYP3A4 inhibitors).4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no human data on the effect of ivosidenib on fertility. No specific fertility studies have been conducted in animals, but undesirable effects on reproductive organs were observed in a 28-day repeat-dose toxicity study in rats. Uterine atrophy was observed in females at non-tolerated dose levels approximately 1.7-fold the clinical exposure (based on AUC) and was reversible after a 14-day recovery period. Testicular degeneration was observed in males at non-tolerated dose levels approximately 1.2-fold the clinical exposure (based on AUC) and reversibility of this finding has not been assessed. The clinical relevance of these effects is unknown.
(Category D)
There are no human data, but based on animal data, Tibsovo may cause fetal harm if administered during pregnancy. Reproductive toxicity (embryofetal mortality and growth alteration) was seen in animal studies, starting at 2-fold the steady-state clinical exposure (based on AUC) at the recommended human dose (see Preclinical data).
Advise patients of the risk to the fetus if Tibsovo is used during pregnancy. Assess pregnancy status prior to starting treatment with Tibsovo. Advise patients to use effective contraception during treatment with Tibsovo and for at least 1 month after the last dose.
As ivosidenib may decrease systemic concentrations of hormonal contraceptives, concomitant use of an alternative contraceptive method such as barrier contraceptives is recommended (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Preclinical data.
In embryofetal development studies in rats, lower fetal body weights, delayed skeletal ossification and development variation of major blood vessels occurred in the absence of maternal toxicity. In rabbits, maternal toxicity, spontaneous abortions, decreased fetal body weights, increased post-implantation loss, delayed skeletal ossification and visceral development variation (small spleen) were observed. In rats and rabbits, the no-adverse-effect levels for embryofetal development were 0.4-fold and 1.4-fold the clinical exposure (based on AUC), respectively. Animal studies indicate that ivosidenib crosses the placenta and is found in fetal plasma. It is not known whether ivosidenib or its metabolites are excreted in milk.
There are no data on the presence of ivosidenib or its metabolites in human milk, the effects on a breastfed child, or the effects on milk production. Due to the potential risk to a breastfed child, breastfeeding should be discontinued during treatment with Tibsovo and for at least one month after the last dose.4.7 Effects on Ability to Drive and Use Machines
Tibsovo does not have sedating properties and does not specifically change the ability to drive and use machines. However, any adverse effects of Tibsovo which a patient experiences (which could include fatigue, dizziness and QTc interval prolongation - see Section 4.8 Adverse Effects (Undesirable Effects)) should be considered when assessing ability to drive or operate machines.
4.8 Adverse Effects (Undesirable Effects)
Safety profile in cholangiocarcinoma.
The safety profile of Tibsovo was studied in 123 patients with previously treated, locally advanced or metastatic cholangiocarcinoma in Study AG120-C-005. Patients received at least one dose of either Tibsovo 500 mg daily (n=123) or placebo (n=59).
The median (range) and mean (standard deviation, SD) duration of treatment with Tibsovo were 2.8 (0.1 to 45.1) months and 6.7 (8.2) months, respectively.
Serious adverse events occurred in 35% of patients receiving Tibsovo. Serious adverse events that occurred in ≥ 2% of patients in the Tibsovo arm were pneumonia, ascites, hyperbilirubinaemia, and jaundice cholestatic. Fatal adverse events occurred in 4.9% of patients receiving Tibsovo, including sepsis (1.6%) and pneumonia, intestinal obstruction, pulmonary embolism, and hepatic encephalopathy (each 0.8%).
Tibsovo was permanently discontinued in 7% of patients. The most common adverse event leading to permanent discontinuation was acute kidney injury (1.6%).
Dose interruptions due to adverse events occurred in 30% of patients treated with Tibsovo. The most common (> 2%) adverse events leading to dose interruption were hyperbilirubinaemia, alanine aminotransferase increased, aspartate aminotransferase increased, ascites, and fatigue.
Dose reductions of Tibsovo due to an adverse event occurred in 4% of patients. Adverse events leading to dose reduction were electrocardiogram QTc interval prolongation (3.3%) and neuropathy peripheral (0.8%).
The most common adverse events and laboratory abnormalities in patients who received Tibsovo in Study AG120-C-005 are presented in Table 3 and Table 4, respectively.


Safety profile in acute myeloid leukaemia (AML).
Summary of the safety profile in AML across studies. Tibsovo 500 mg daily has been studied in combination with azacitidine in newly diagnosed AML (Study AG120-C-009 [N=71]) and as monotherapy in patients with newly diagnosed or with relapsed/refractory AML (Study AG120-C-001 [N=213]). These studies are described further in Section 5.1 Pharmacodynamic Properties. In patients with AML across both of these studies, the most common adverse events including laboratory abnormalities (≥ 25% in either trial) were leukocytes decreased, diarrhoea, haemoglobin decreased, platelets decreased, glucose increased, fatigue, alkaline phosphatase increased, oedema, potassium decreased, nausea, vomiting, phosphatase decreased, decreased appetite, sodium decreased, leukocytosis, magnesium decreased, aspartate aminotransferase increased, arthralgia, dyspnoea, uric acid increased, abdominal pain, creatinine increased, mucositis, rash, electrocardiogram QTc interval prolongation, differentiation syndrome, calcium decreased, neutrophils decreased, and myalgia.
Safety profile in the treatment of newly diagnosed AML (Tibsovo in combination with azacitidine).
In the randomised, phase 3 Study AG120-C-009, patients with newly diagnosed AML received azacitidine in combination with either Tibsovo (500 mg daily, n=72) or matching placebo (n=74). The median duration of treatment with Tibsovo was 7.8 months (range: 0.1 to 40.0 months). Forty patients (55.6%) were exposed to Tibsovo for at least six months and twenty-nine patients (40.3%) were exposed for at least one year.
The most common adverse events and laboratory abnormalities reported in patients who received Tibsovo and azacitidine are shown in Table 5 and Table 6, respectively.
The most common (≥ 5%) serious adverse event to Tibsovo was differentiation syndrome (8%). There were eleven fatal adverse events in patients who received Tibsovo (15%) - most common (≥ 2%) included pneumonia (3%) and haemorrhage intracranial (3%).
The frequency of discontinuation of Tibsovo due to adverse events was 29%. Adverse event leading to discontinuation of Tibsovo in ≥ 2% of patients was pulmonary embolism (3%).
The frequency of dose interruption of Tibsovo due to adverse events was 67%. The most common (≥ 5%) adverse events leading to dose interruption of Tibsovo were neutropenia (24%), febrile neutropenia (13%), pneumonia (13%), thrombocytopenia (7%) and electrocardiogram QTc interval prolongation (7%).
The frequency of dose reduction of Tibsovo due to adverse events was 22%. The most common (≥ 5%) adverse events leading to dose reduction were electrocardiogram QTc interval prolongation (10%) and neutropenia (8%).


Safety profile in the treatment of newly diagnosed AML (Tibsovo monotherapy).
The safety profile of single-agent Tibsovo at a dose of 500 mg daily was studied in 28 adults with newly diagnosed AML in a single-arm, open-label, multicenter clinical trial (Study AG120-C-001).
The median duration of exposure to Tibsovo amongst this group was 4.3 months (range: 0.3 to 40.9 months). Ten patients (36%) were exposed to Tibsovo for at least six months and six patients (21%) were exposed for at least one year.
Common (≥ 5%) serious adverse events included differentiation syndrome (18%), electrocardiogram QTc interval prolongation (7%), and fatigue (7%). There was one case of posterior reversible encephalopathy syndrome (PRES).
Common (≥ 10%) adverse events leading to dose interruption included electrocardiogram QTc interval prolongation (14%) and differentiation syndrome (11%). Two (7%) patients required a dose reduction due to electrocardiogram QTc interval prolongation. One patient each required permanent discontinuation due to diarrhoea and PRES.
The most common adverse events and changes in selected post-baseline laboratory values reported in Study AG120-C-001 amongst these patients with newly diagnosed AML are shown in Table 7 and Table 8, respectively.


Safety profile in the treatment of relapsed or refractory AML (Tibsovo monotherapy).
The safety profile of single-agent Tibsovo was studied in 179 adults with relapsed or refractory AML who received Tibsovo 500 mg daily in Study AG120-C-001 (see Section 5.1 Pharmacodynamic Properties).
The median duration of exposure to Tibsovo was 3.9 months (range: 0.1 to 39.5 months). Sixty-five patients (36%) were exposed to Tibsovo for at least six months and 16 patients (9%) were exposed for at least 1 year.
Serious treatment emergent adverse events (TEAEs) (≥ 5%) were differentiation syndrome (10%), leukocytosis (10%), and electrocardiogram QTc interval prolongation (7%). There was one case of progressive multifocal leukoencephalopathy (PML).
The most common adverse events leading to dose interruption were electrocardiogram QTc interval prolongation (7%), differentiation syndrome (3%), leukocytosis (3%) and dyspnoea (3%). Five out of 179 patients (3%) required a dose reduction due to an adverse reaction. Adverse events leading to a dose reduction included electrocardiogram QTc interval prolongation (1%), diarrhoea (1%), nausea (1%), decreased haemoglobin (1%), and increased transaminases (1%). Adverse events leading to permanent discontinuation included Guillain-Barré syndrome (1%), rash (1%), stomatitis (1%), and creatinine increased (1%).
The most common adverse events and changes in selected post-baseline laboratory values reported in this group of patients with relapsed or refractory AML are shown in Table 9 and Table 10, respectively.
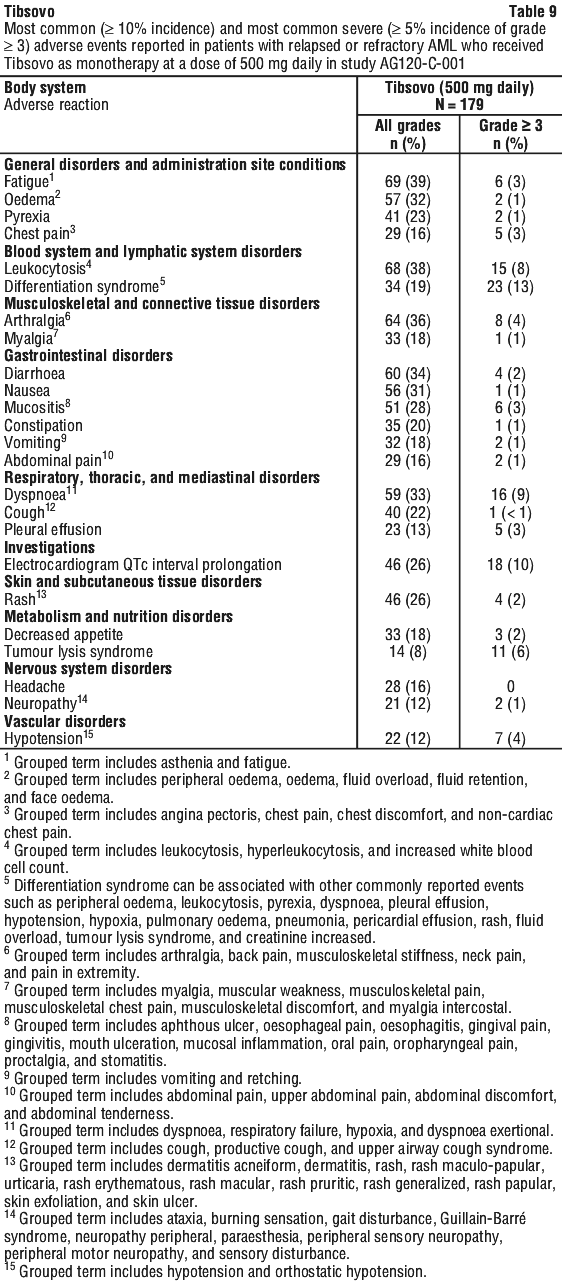

Description of selected adverse events.
Differentiation syndrome in patients with AML.
Differentiation syndrome is a known risk associated with Tibsovo treatment in patients with AML (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
In the monotherapy clinical trial AG120-C-001, 25% (7/28) of patients with newly diagnosed AML and 19% (34/179) of patients with relapsed or refractory AML treated with Tibsovo experienced differentiation syndrome. Of the seven patients with newly diagnosed AML who experienced differentiation syndrome, six (86%) patients recovered. Of the 34 patients with relapsed or refractory AML who experienced differentiation syndrome, 27 (79%) patients recovered after treatment or after dose interruption of Tibsovo. Differentiation syndrome occurred as early as one day and up to three months after initiation with Tibsovo and has been observed with or without concomitant leukocytosis.
In Study AG120-C-009, 14% (10/71) of patients with newly diagnosed AML treated with Tibsovo in combination with azacitidine experienced differentiation syndrome. Of these ten patients, two required dose interruptions to manage signs/symptoms and zero patients permanently discontinued Tibsovo treatment due to differentiation syndrome. The median time to onset of differentiation syndrome after initiation of combination therapy was 20 days, with a range of three to 46 days.
QTc interval prolongation.
Prolongation of the electrocardiogram QTc interval is a known risk associated with Tibsovo treatment and may occur at any time during treatment (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
In Study AG120-C-005, in the 123 patients with locally advanced or metastatic cholangiocarcinoma treated with Tibsovo monotherapy, QTc prolongation was reported in 10%; 2% experienced Grade 3 or higher reactions. Based on the analysis of the ECGs, 2% of patients had a QTc interval > 500 msec and 5% had QTc interval prolongation > 60 msec from baseline. No patient discontinued treatment due to QTc prolongation, and dose reduction to manage signs/symptoms was required in (3% of patients). The median time to onset after treatment initiation was 28 days (range: one day to 698 days [23 months]).
Of the 258 patients with hematological malignancies treated with Tibsovo monotherapy in the clinical trial AG120-C-001, 9% were found to have a QTc interval greater than 500 msec and 14% of patients had an increase from baseline QTc greater than 60 msec (see Section 4.8 Adverse Effects (Undesirable Effects)). One patient developed ventricular fibrillation attributed to Tibsovo. The clinical trial excluded patients with baseline QTc of ≥ 450 msec (unless the QTc ≥ 450 msec was due to a pre-existing bundle branch block) or with a history of long QT syndrome or uncontrolled or significant cardiovascular disease.
In Study AG120-C-009, in the 71 patients with newly diagnosed AML treated with Tibsovo in combination with azacitidine, electrocardiogram QT prolonged was reported in 20%; 10% experienced Grade 3 or higher reactions. Based on the analysis of the ECGs, 14% of patients treated with Tibsovo in combination with azacitidine, who had at least one post-baseline ECG assessment, were found to have a QTc interval > 500 msec, 22% had an increase from baseline QTc ˃ 60 msec. One patient discontinued Tibsovo treatment due to electrocardiogram QT prolongation. Dose interruption and reduction were required in 6% and 9% of patients, respectively. The median time to onset of QT prolongation in patients treated with Tibsovo was 29 days. Electrocardiogram QT prolongation occurred as early as 1 day and up to 5 months after treatment initiation.
Guillain-Barré syndrome.
Two cases of Guillain-Barré syndrome (< 1%) occurred in patients with haematological malignancies receiving Tibsovo in Study AG120-C-001. There were no cases in Study AG120-C-009 or Study AG120-C005.
Special populations.
Elderly.
Of the 72 patients with newly diagnosed AML treated with Tibsovo in combination with azacitidine, 94% were 65 years of age or older, and 54% were 75 years or older. Of the 34 patients with newly diagnosed AML treated with Tibsovo monotherapy, 97% were 65 years of age or older, and 56% were 75 years or older. Of the 179 patients with relapsed or refractory AML treated with Tibsovo monotherapy, 63% were 65 years of age or older and 22% were 75 years or older. Of 123 patients with cholangiocarcinoma treated with Tibsovo in Study AG120-C-005, 36% were ≥ 65 years of age and 11% were ≥ 75 years of age. No overall difference in safety was observed between patients ≥ 65 years old and younger patients.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
In the event of overdose, toxicity may manifest as QTc interval prolongation or ventricular arrhythmia, or exacerbation of other adverse reactions (see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)). Patients should be closely monitored and provided with appropriate supportive care (see Section 4.2 Dose and Method of Administration). There is no specific antidote for Tibsovo overdose.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antineoplastic agents; other antineoplastic agents. ATC code: L01XX62.
Mechanism of action.
Ivosidenib is a small molecule inhibitor of certain mutant isocitrate dehydrogenase 1 (IDH1) enzymes. Through a gain of neomorphic function, the mutant IDH1 converts alpha-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG). As 2-HG competitively inhibits α-KG-dependent enzymes, including histone and DNA demethylases, its accumulation leads to widespread epigenetic dysregulation.
Ivosidenib inhibited selected IDH1 mutants (R132C, R132L, R132G, R132H and R132S) at much lower concentrations than wild-type IDH1 in vitro.
Cholangiocarcinoma.
In a patient-derived xenograft intra-hepatic cholangiocarcinoma mouse model with IDH1 R132C, ivosidenib reduced 2-HG levels.
Acute myeloid leukaemia.
Inhibition of the mutant IDH1 enzyme by ivosidenib led to decreased 2-HG levels and induced myeloid differentiation in vitro and in vivo in mouse xenograft models of IDH1-mutated AML. In blood samples from patients with AML with mutated IDH1, ivosidenib decreased 2-HG levels, reduced blast counts, and increased percentages of mature myeloid cells.
Pharmacodynamic effects.
Multiple doses of ivosidenib 500 mg daily decreased plasma concentrations of 2-HG in patients with haematological malignancies and cholangiocarcinoma with mutated IDH1 to levels approximating those observed in healthy subjects. In tumour biopsies from patients with cholangiocarcinoma who received ivosidenib, the mean (coefficient of variation [CV]) reduction in 2-HG concentrations was 82% (32%). In bone marrow biopsies from patients with haematological malignancies who received ivosidenib, the mean (CV) reduction in 2-HG concentrations was 93% (11%).
Concentration-dependent prolongation of the QTc interval was observed following administration of ivosidenib at the recommended dose in patients with haematological malignancies and solid tumours. The mean maximal prolongation in both settings was 17 msec, with an upper confidence interval of 20 msec. Co-administration of moderate or strong CYP3A inhibitors is expected to further increase QTc interval prolongation from baseline. See Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use.
Clinical trials (efficacy).
Efficacy in previously treated, locally advanced or metastatic cholangiocarcinoma.
The efficacy of Tibsovo was evaluated in a randomised (2:1), multicentre, double-blind, placebo-controlled, phase 3 clinical trial (Study AG120-C-005, also known as 'ClarIDHy') of 185 adult patients with locally advanced or metastatic cholangiocarcinoma with an IDH1 mutation whose disease had progressed following at least 1 but not more than 2 prior treatment regimens, including at least 1 gemcitabine- or 5-FU-containing regimen. Patients with certain IDH1 mutations (R132C, R132CL, R132G, R132H or R132S) were selected using a central diagnostic next generation sequencing assay (the Oncomine Focus Assay).
Patients were randomised to receive either Tibsovo 500 mg orally once daily or matched placebo until disease progression or development of unacceptable toxicity. Randomisation was stratified by number of prior therapies (1 or 2). Eligible patients who were randomised to placebo were allowed to cross over to receive Tibsovo after documented radiographic disease progression as assessed by the Investigator.
Tumour imaging assessments were performed every 6 weeks for the first 8 assessments and every 8 weeks thereafter. The primary efficacy outcome was progression-free survival (PFS) assessed by an independent review committed (IRC) according to response evaluation criteria in solid tumors (RECIST) v1.1.
The median age was 62 years (range: 33 to 83). Most patients were female (63%), 57% were Caucasian, all patients had an ECOG performance status of 0 (37%) or 1 (62%), and 47% had received two prior lines of systemic therapy. Most patients had intrahepatic cholangiocarcinoma (91%) at diagnosis and 92% had metastatic disease. Across both arms, 70% of patients had an R132C mutation, 15% had an R132L mutation, 12% had an R132G mutation, 1.6% had an R132S mutation, and 1.1% had an R132H mutation. No patients with R132H-mutant IDH1 were randomised to Tibsovo.
The study demonstrated a statistically significant improvement in PFS. The efficacy results for Study AG120-C-005 are summarised in Table 11 and Figure 1.


Efficacy in newly diagnosed AML - Tibsovo in combination with azacitidine.
The efficacy and safety of Tibsovo was evaluated in a randomised, multicentre, double-blind, placebo-controlled clinical trial (Study AG120-C-009) of 146 adult patients with newly diagnosed AML with an IDH1 mutation who were 75 years or older, or had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline Eastern Cooperative Oncology Group (ECOG) performance status of 2, severe cardiac or pulmonary disease, hepatic impairment with bilirubin > 1.5 times the upper limit of normal, creatinine clearance < 45 mL/min, or other comorbidity. IDH1 mutations were confirmed centrally using the Abbott RealTime IDH1 Assay. Local diagnostic tests were permitted for screening and randomisation, provided a bone marrow or peripheral blood sample was sent for central confirmation. Gene mutation analysis to document IDH1 mutated disease from a bone marrow or peripheral blood sample was conducted for all patients. Patients were randomised to receive either Tibsovo 500 mg, or matched placebo, orally once daily on Days 1-28, in combination with azacitidine 75 mg/m2/day subcutaneously or intravenously on Days 1-7 or Days 1-5 and 8-9 of each 28-day cycle. Treatment was continued for a minimum of 6 cycles unless they experienced disease progression or unacceptable toxicity or underwent haematopoietic stem cell transplantation.
The primary efficacy outcome was event-free survival (EFS), measured from the date of randomisation until treatment failure, relapse from remission, or death by any cause. Treatment failure was defined as failure to achieve complete remission (CR) by week 24. Overall Survival (OS), CR rate, CR + CR with partial hematologic recovery (CR + CRh) rate and objective response rate (ORR) were key secondary efficacy endpoints.
Amongst patients randomised to receive Tibsovo, the median age was 76 years (range: 58 to 84); 58% were male; 21% Asian and 17% were Caucasian, whilst ethnicity was not reported for 61%. ECOG performance status was 0 (19%), 1 (44%), or 2 (36%), and most patients (75%) had de novo AML. Cytogenetic risk (per National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology, 2017), was intermediate for most patients (67%), with 4% having favourable and 26% having poor/other cytogenetic risk. According to the central testing, 63% of patients had an R132C mutation, 19% had an R132H mutation, 8% had an R132G mutation, 4% had an R132L mutation, and 3% had an R132H mutation.
The key efficacy findings of Study AG120-C-009 are summarised in Table 12 and Figure 2.

 The median time to first CR for Tibsovo with azacitidine was four months (range: 1.7 to 9.2 months). The median time to first CR + CRh for Tibsovo with azacitidine was four months (range: 1.7 to 8.6 months). The median time to first objective response (defined as CR, CRi (including CRp), PR or MLFS) was two months (range: 1.7 to 7.5 months) for Tibsovo with azacitidine.
The median time to first CR for Tibsovo with azacitidine was four months (range: 1.7 to 9.2 months). The median time to first CR + CRh for Tibsovo with azacitidine was four months (range: 1.7 to 8.6 months). The median time to first objective response (defined as CR, CRi (including CRp), PR or MLFS) was two months (range: 1.7 to 7.5 months) for Tibsovo with azacitidine.
Efficacy in newly diagnosed AML - Tibsovo monotherapy.
The efficacy of Tibsovo was evaluated in an open-label, single-arm, multicentre clinical trial (Study AG120C-001) that included 28 adult patients with newly diagnosed AML with an IDH1 mutation. IDH1 mutations were identified by a local or central diagnostic test and confirmed retrospectively using the Abbott RealTime IDH1 Assay. The cohort included patients who were age 75 years or older or who had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline Eastern Cooperative Oncology Group (ECOG) performance status of ≥ 2, severe cardiac or pulmonary disease, hepatic impairment with bilirubin > 1.5 times the upper limit of normal, or creatinine clearance 45 mL/min. Tibsovo was given orally at a starting dose of 500 mg daily until disease progression, development of unacceptable toxicity, or undergoing haematopoietic stem cell transplantation. Two (7%) of the 28 patients went on to stem cell transplantation following Tibsovo treatment.
Amongst this group of patients, the median age was 77 years (range: 64 to 87). Most patients were male (54%), 87% were Caucasian, ECOG performance status was 0 in 37%, 1 in 62%, two in five patients (18%) and 3 in one patient (4%). Just under half (46%) had received a hypomethylating agent previously for an antecedent haematological disorder. Most patients (61%) were transfusion dependent at baseline, defined as receipt of any transfusion within 56 days prior to the first dose of Tibsovo. Most patients had AML with myelodysplasia-related changes (68%), whilst 21% had de novo AML and 11% had therapy-related AML. European Leukaemia Net risk category was adverse for most patients (68%) and intermediate for the remaining 32%. According to the central retrospective confirmatory testing, 86% of patients had an R132C mutation, 7% had an R132H mutation, there was 1 patient each with an R132G or R132L mutation, and no patients with an R132S mutation.
Efficacy was established on the basis of the rate of complete remission (CR) or complete remission with partial hematologic recovery (CRh), the duration of CR + CRh, and the rate of conversion from transfusion dependence to transfusion independence. The efficacy results are shown in Table 13. The median follow-up was 8.1 months (range: 0.6 to 40.9 months) and median treatment duration was 4.3 months (range: 0.3 to 40.9 months).
 For patients who achieved a CR or CRh, the median time to CR or CRh was 2.8 months (range: 1.9 to 12.9 months). Of the 12 patients who achieved a best response of CR or CRh, 11 (92%) achieved a first response of CR or CRh within six months of initiating Tibsovo.
For patients who achieved a CR or CRh, the median time to CR or CRh was 2.8 months (range: 1.9 to 12.9 months). Of the 12 patients who achieved a best response of CR or CRh, 11 (92%) achieved a first response of CR or CRh within six months of initiating Tibsovo.
Among the 17 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, seven (41.2%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 11 patients who were independent of both RBC and platelet transfusions at baseline, six (54.5%) remained transfusion independent during any 56-day post-baseline period.
Efficacy in relapsed or refractory AML.
The efficacy of Tibsovo was evaluated in an open-label, single-arm, multicenter clinical trial (Study AG120C-001) in 174 adult patients with relapsed or refractory AML with an IDH1 mutation. IDH1 mutations were identified by a local or central diagnostic test and confirmed retrospectively using the Abbott RealTime IDH1 Assay. Tibsovo was given orally at a starting dose of 500 mg daily until disease progression, development of unacceptable toxicity, or undergoing haematopoietic stem cell transplantation. Twenty-one (12%) of the 174 patients went on to stem cell transplantation following Tibsovo treatment.
Amongst this group of patients, the median age was 67 years (range: 18 to 87). Half of the group were male (51%), 62% were Caucasian, ECOG performance status was 0 in 21%; 1 in 56%; 2 in 22% and 3 in two patients (1%). Most patients had de novo AML (67%) and 33% had secondary AML. Relapse was primary refractory for 37%, untreated relapse for 37% and refractory relapse for 26% of patients. Most patients (63%) were transfusion dependent at baseline (defined as receipt of any transfusion within 56 days prior to the first dose of Tibsovo), and the median number of prior therapies was 2 (range: 1-6): 23% had received prior stem cell transplantation for AML. Cytogenetic risk was intermediate for most patients (60%), with 27% having poor cytogenetic risk and the remainder unknown. According to the central retrospective confirmatory testing, 59% of patients had an R132C mutation, 25% had R132H, 7% had R132G, 6% had R132S, and 4% had an R132L mutation.
Efficacy was established on the basis of the rate of complete remission (CR) plus complete remission with partial haematologic recovery (CRh), the duration of CR + CRh, and the rate of conversion from transfusion dependence to transfusion independence. The efficacy results are shown in Table 14. The median follow-up was 8.3 months (range: 0.2 to 39.5 months) and median treatment duration was 4.1 months (range: 0.1 to 39.5 months).
 For patients who achieved a CR or CRh, the median time to CR or CRh was two months (range: 0.9 to 5.6 months). Of the 57 patients who achieved a best response of CR or CRh, all achieved a first response of CR or CRh within six months of initiating Tibsovo.
For patients who achieved a CR or CRh, the median time to CR or CRh was two months (range: 0.9 to 5.6 months). Of the 57 patients who achieved a best response of CR or CRh, all achieved a first response of CR or CRh within six months of initiating Tibsovo.
Among the 110 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 41 (37%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 64 patients who were independent of both RBC and platelet transfusions at baseline, 38 (59%) remained transfusion independent during any 56-day post-baseline period.
5.2 Pharmacokinetic Properties
A summary of ivosidenib pharmacokinetic (PK) parameters following administration of ivosidenib 500 mg as a single or daily dose (for steady state) is provided in Table 15. The AUC and Cmax of ivosidenib increase in a less than dose proportional manner from 200 mg to 1,200 mg once daily (0.4 to 2.4 times the recommended dose). Steady-state PK is reached within 14 days with daily dosing.

Metabolism.
Ivosidenib was the predominant component (> 92%) of total radioactivity in plasma from healthy subjects. It is primarily metabolised by CYP3A4 with minor contributions by N-dealkylation and hydrolytic pathways.
Special populations.
No clinically meaningful effects on the pharmacokinetics of ivosidenib were observed based on age (18 to 89 years), sex, race, body weight (38 to 150 kg), ECOG performance status, mild or moderate renal impairment (eGFR ≥ 30 mL/min/1.73 m2), and mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment. The pharmacokinetics of ivosidenib in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2), including patients requiring dialysis, and in patients with severe hepatic impairment (Child-Pugh class C) are unknown.
5.3 Preclinical Safety Data
Genotoxicity.
Ivosidenib was not mutagenic in a bacterial reverse mutation assay or clastogenic in vitro in human lymphocytes or in vivo in a rat micronucleus assay.
Carcinogenicity.
Carcinogenicity studies have not been conducted with ivosidenib.6 Pharmaceutical Particulars
6.1 List of Excipients
Microcrystalline cellulose, croscarmellose sodium, hypromellose acetate succinate, colloidal anhydrous silica, magnesium stearate, sodium lauryl sulfate, hypromellose, titanium dioxide, lactose monohydrate, triacetin, indigo carmine aluminium lake.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store in a dry place below 30°C. Keep the bottle tightly closed to protect from moisture.
6.5 Nature and Contents of Container
White, high-density polyethylene (HDPE) bottle with polypropylene (PP) child resistant closure and polyethylene (PE) faced induction heat seal liner. Each bottle contains 60 film-coated tablets and a silica gel desiccant in a HDPE canister. The bottles are packaged in a cardboard box; each box contains 1 bottle.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
The active component of Tibsovo is ivosidenib which is a white to light yellow solid and has the chemical name: Glycinamide, 1-(4-cyano-2-pyridinyl)-5-oxo-L-prolyl-2-(2-chlorophenyl)-N-(3,3-difluorocyclobutyl)-N2-(5-fluoro-3-pyridinyl)-, (2S)- and molecular formula: C28H22ClF3N6O3 (MW = 583.0). Ivosidenib is practically insoluble in aqueous solutions and is variably soluble in various organic solvents.
Chemical structure.
The chemical structure of ivosidenib free form drug substance:

CAS number.
1448347-49-6.7 Medicine Schedule (Poisons Standard)
S4 - Prescription only medicine.
Summary Table of Changes
