

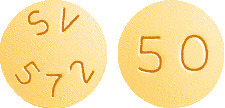
What is in this leaflet?
Please read this leaflet carefully before you take TIVICAY. This leaflet answers some common questions about TIVICAY (dolutegravir). It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the expected benefits of you taking TIVICAY against the risks this medicine could have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What is TIVICAY used for?
TIVICAY is used to treat HIV (human immunodeficiency virus) infection in adults and in children over 6 years old.
TIVICAY does not cure HIV infection; it reduces the amount of virus in your body and keeps it at a low level. TIVICAY also increases the CD4 cell count in your blood. CD4 cells are a type of white blood cells that are important in helping your body to fight infection.
TIVICAY is used, in combination with other anti-retroviral medicines (combination therapy), to treat HIV infection in adults and children over 6 years old. To control your HIV infection, and to stop your illness from getting worse, you must keep taking all your medicines, unless your doctor tells you to stop taking any.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
TIVICAY is not addictive.
There is not enough information to recommend the use of this medicine for children under the age of 6 years.
This medicine is available only with a doctor's prescription.
Before you take TIVICAY
When you must not take it
You must not take TIVICAY if:
- you're taking another medicine called dofetilide, pilsicainide (used to treat heart conditions) or fampridine (used in multiple sclerosis).
Do not take TIVICAY if you have an allergy to:
- any medicine containing dolutegravir
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
If you think any of these apply to you, don't take TIVICAY until you have checked with your doctor.
Tell your doctor if you:
- have liver problems, including hepatitis B or C.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Some other conditions may develop during HIV treatment.
Symptoms of infection and inflammation
People with advanced HIV infection (AIDS) have weak immune systems and are more likely to develop serious infections (opportunistic infections). When they start treatment, the immune system becomes stronger, so the body starts to fight infections.
Symptoms of infection and inflammation may develop, caused by either:
- old, hidden infections flaring up again as the body fights them
- the immune system attacking healthy body tissue (autoimmune disorders)
The symptoms of autoimmune disorders may develop many months after you start taking medicine to treat your HIV infection.
Symptoms may include:
- muscle weakness and/or muscle pain
- joint pain or swelling
- weakness beginning in the hands and feet and moving up towards the trunk of the body
- palpitations or tremor
- hyperactivity (excessive restlessness and movement).
If you get any symptoms of infection or if you notice any of the symptoms above:
Tell your doctor immediately. Don't take other medicines for the infection without your doctor's advice.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
If you are pregnant, or think you could be, or if you are planning to have a baby, don't take TIVICAY without checking with your doctor.
Your doctor will consider the benefit to you and the risk to your baby of taking TIVICAY while you're pregnant.
If you could get pregnant while taking TIVICAY, you need to use a reliable method of contraception to prevent pregnancy. Your doctor can discuss with you the risks and benefits involved.
Taking TIVICAY at the time of becoming pregnant, or during the fist twelve weeks of pregnancy, may increase the risk of a type of birth defect, called neural tube defect, such as spina bifida (malformed spinal cord).
Where possible, women who are HIV-positive should not breast feed, because HIV infection can be passed on to the baby in breast milk.
It is not known whether the ingredients of TIVICAY can pass into breast milk and harm your baby.
If you have not told your doctor about any of the above, tell him/her before you start taking TIVICAY.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Don't take TIVICAY with this medicine:
- dofetilide or pilsicainide, to treat heart conditions
- fampridine (used in multiple sclerosis)
Tell your doctor if you are taking any of the medicines in the following list:
- metformin, to treat diabetes
- medicines called antacids, to treat indigestion and heartburn.
- calcium and iron supplements.
- etravirine, efavirenz, fosamprenavir/ritonavir, nevirapine or tipranavir/ritonavir, to treat HIV infection
- rifampicin, to treat tuberculosis (TB) and other bacterial infections
- phenytoin and phenobarbital, to treat epilepsy
- carbamazepine, to treat epilepsy and bipolar disorder
- St. John's wort, (Hypericum perforatum), a herbal remedy to treat depression
These medicines may be affected by TIVICAY or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How do I take TIVICAY?
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
Adults
- The usual dose of TIVICAY is one 50 mg tablet, once a day; or
- For HIV infection that is resistant to other medicines similar to TIVICAY, the usual dose of TIVICAY is one 50 mg tablet, twice a day.
Your doctor will decide on the correct dose of TIVICAY for you.
Children
Your doctor will decide on the correct dose of TIVICAY for your child, depending on the weight of the child.
How to take it
Swallow the tablets whole with a full glass of water.
Antacid medicines
Antacids, to treat indigestion and heartburn, can stop TIVICAY being absorbed into your body and make it less effective.
Do not take an antacid during the 6 hours before you take TIVICAY, or for at least 2 hours after you take it. Other acid-lowering medicines like ranitidine and omeprazole can be taken at the same time as TIVICAY.
Talk to your doctor for further advice on taking acid-lowering medicines with TIVICAY.
Calcium or iron supplements
Calcium or iron supplements can stop TIVICAY being absorbed into your body and make it less effective.
Do not take a calcium or iron supplement during the 6 hours before you take TIVICAY, or for at least 2 hours after you take it. If you take food with TIVICAY, then you can take calcium and iron supplements at the same time as TIVICAY.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it for
Continue taking your medicine for as long as your doctor tells you.
If you forget to take it
If you miss a dose, take it as soon as you remember. But if it is less than 4 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (In Australia call 131126. In New Zealand call 0800 POISON or 0800 764 766) for advice if you think you or anyone else may have taken too much TIVICAY, even if there are no signs of discomfort or poisoning. If you are not sure what to do, contact your doctor or pharmacist. You may need urgent medical attention.
While you are using TIVICAY
You will need regular blood tests For as long as you're taking TIVICAY, your doctor will arrange regular blood tests to check for side effects.
Stay in regular contact with your doctor TIVICAY helps to control your condition, but it is not a cure for HIV infection. You need to keep taking it every day to stop your illness from getting worse. Because TIVICAY does not cure HIV infection, you may still develop other infections and illnesses linked to HIV infection.
Keep in touch with your doctor, and don't stop taking TIVICAY without your doctor's advice.
Protect other people
HIV infection is spread by sexual contact with someone who has the infection, or by transfer of infected blood (for example, by sharing injection needles). You can still pass on HIV when taking this medicine, although the risk is lowered by effective antiretroviral therapy. Discuss with your doctor the precautions needed to avoid infecting other people, which may include:
- Use a condom when you have oral or penetrative sex.
- Don't risk blood transfer - for example, don't share needles.
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking TIVICAY.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
TIVICAY can make you dizzy and have other side effects that make you less alert.
Don't drive or use machines unless you are sure you're not affected.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TIVICAY.
This medicine helps most people with HIV, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
When you're being treated for HIV, it can be hard to tell whether a symptom is a side effect of TIVICAY or other medicines you are taking, or an effect of the HIV disease itself. So it is very important to talk to your doctor about any changes in your health.
Some side effects may only be seen in your blood tests and may not appear immediately after you start taking TIVICAY. If you get any of these effects, and if they are severe, your doctor may advise you to stop taking TIVICAY.
As well as the effects listed below for TIVICAY, other conditions can develop during combination therapy for HIV.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Allergic reactions
- See a doctor as soon as possible if you develop a rash.
Allergic reactions are uncommon in people taking TIVICAY. Signs include:
- skin rash
- a high temperature (fever)
- lack of energy (fatigue)
- swelling, sometimes of the face or mouth (angioedema), causing difficulty in breathing
- muscle or joint aches.
Your doctor may decide to carry out tests on your liver, kidneys or blood, and may tell you to stop taking TIVICAY.
Very common side effects
These may affect more than 1 in 10 people:
- headache
- diarrhoea
- feeling sick (nausea)
Common side effects
These may affect up to 1 in 10 people:
- rash
- itching (pruritus)
- being sick (vomiting)
- stomach (abdominal) pain or discomfort
- difficulty in sleeping (insomnia)
- dizziness
- abnormal dreams
- depression (feelings of deep sadness and unworthiness)
- anxiety
- lack of energy (fatigue)
- wind (flatulence)
Uncommon side effects
These may affect up to 1 in 100 people:
- inflammation of the liver (hepatitis)
- an inflammatory condition which may develop as the immune system becomes stronger (immune reconstitution syndrome or 'IRIS')
- allergic reaction (hypersensitivity) (see earlier in this section for more details)
- suicidal thoughts*
- suicidal attempt*
- joint pain
- muscle pain
- weight gain
* mainly in patients who have had depression or mental health problems before
Rare side effects
These may affect up to 1 in 1000 people:
- liver failure (signs may include yellowing of the skin and the whites of the eyes or unusually dark urine
Other side Effects that may show up in blood tests
Other side effects have occurred in some people but their exact frequency is unknown:
- increase in bilirubin (a substance produced by the liver) in the blood
- an increase in the level of enzymes produced in the muscles (creatine phosphokinase, creatinine)
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using TIVICAY
Storage
Keep your tablets in the bottle until it is time to take them.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
10 mg tablets only - Store in the original package in order to protect from moisture. Keep the bottle tightly closed. Do not remove the desiccant.
Do not store TIVICAY or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
10 mg - White, film-coated, round, biconvex tablets debossed with 'SV 572' on one side and '10' on the other side.
25 mg - Pale yellow, film-coated, round, biconvex tablets debossed with 'SV 572' on one side and '25' on the other side.
50 mg - Yellow, film-coated, round, biconvex tablets debossed with 'SV 572' on one side and '50' on the other side.
TIVICAY is available in bottles of 30 tablets with child-resistant closure.
The 10 mg tablet bottles contain a desiccant to reduce moisture. Once the bottle has been opened, keep the desiccant in the bottle, do not remove it.
Ingredients
TIVICAY contains 10 mg, 25 mg or 50 mg of dolutegravir (as dolutegravir sodium) as the active ingredient.
- mannitol
- microcrystalline cellulose
- povidone
- sodium starch glycolate Type A
- sodium stearyl fumarate
- polyvinyl alcohol
- titanium dioxide
- macrogol 3350
- talc
- iron oxide yellow (25 mg and 50 mg tablets only)
Supplier
ViiV Healthcare Pty Ltd
Level 4, 436 Johnston St,
Abbotsford, Victoria, 3067
Australia.
Where to go for further information:
Pharmaceutical companies are not in a position to give people an individual diagnosis or medical advice. Your doctor or pharmacist is the best person to give you advice on the treatment of your condition. You may also be able to find general information about your disease and its treatment from patient information groups and product specific organisations.
This leaflet was prepared on 30 October 2019.
Version 10.0
TIVICAY:
10 mg - AUST R 312782
25 mg - AUST R 312781
50mg - AUST R 205212
Trademarks are owned by or licensed to the ViiV Healthcare group of companies.
© 2019 ViiV Healthcare group of companies or its licensor.
Published by MIMS February 2025
 To reduce the risk of choking, do not swallow more than one tablet at a time, and where possible, children weighing 14 to less than 20 kg should preferentially take the dispersible tablet formulation.
To reduce the risk of choking, do not swallow more than one tablet at a time, and where possible, children weighing 14 to less than 20 kg should preferentially take the dispersible tablet formulation. If swallowing the dispersible tablets whole with water, do not swallow more than one tablet at a time to reduce the risk of choking. There are insufficient safety and efficacy data available to recommend a dose for dolutegravir dispersible tablets in children below age 4 weeks or weighing less than 3 kg.
If swallowing the dispersible tablets whole with water, do not swallow more than one tablet at a time to reduce the risk of choking. There are insufficient safety and efficacy data available to recommend a dose for dolutegravir dispersible tablets in children below age 4 weeks or weighing less than 3 kg.

 Laboratory abnormalities with a worsening grade from baseline in ≥ 2% (for grades 3 to 4 combined) of patients are presented in Table 5. Side by side tabulation is to simplify presentation; direct comparisons across trials should not be made due to differing trial designs.
Laboratory abnormalities with a worsening grade from baseline in ≥ 2% (for grades 3 to 4 combined) of patients are presented in Table 5. Side by side tabulation is to simplify presentation; direct comparisons across trials should not be made due to differing trial designs. In a multicentre, open label trial (FLAMINGO), 243 subjects received Tivicay 50 mg once daily versus 242 subjects who received darunavir 800 mg/ ritonavir 100 mg once daily, both in combination with investigator selected NRTI background regimen (either ABC/3TC or TDF/FTC). There were 484 subjects included in the efficacy and safety analyses. Through 48 weeks, the rates of adverse events leading to discontinuation were 2% in subjects receiving Tivicay and 4% in subjects receiving darunavir/ ritonavir. The ADRs observed in FLAMINGO were generally consistent with those seen in SPRING-2 and SINGLE.
In a multicentre, open label trial (FLAMINGO), 243 subjects received Tivicay 50 mg once daily versus 242 subjects who received darunavir 800 mg/ ritonavir 100 mg once daily, both in combination with investigator selected NRTI background regimen (either ABC/3TC or TDF/FTC). There were 484 subjects included in the efficacy and safety analyses. Through 48 weeks, the rates of adverse events leading to discontinuation were 2% in subjects receiving Tivicay and 4% in subjects receiving darunavir/ ritonavir. The ADRs observed in FLAMINGO were generally consistent with those seen in SPRING-2 and SINGLE. In both SPRING-2 and SINGLE studies virologic suppression (HIV-1 RNA < 50 copies/mL), treatment differences were comparable across baseline characteristics (gender, race and age).
In both SPRING-2 and SINGLE studies virologic suppression (HIV-1 RNA < 50 copies/mL), treatment differences were comparable across baseline characteristics (gender, race and age).

 After the monotherapy phase, patients had the opportunity to re-optimise their background regimen when possible.
After the monotherapy phase, patients had the opportunity to re-optimise their background regimen when possible. The response rate at week 48 was sustained with 116/183 (63% [95% CI: 56%, 70%]) subjects having HIV-1 RNA < 50 copies/mL (ITT-E, snapshot algorithm). Response was also sustained through week 48 in subjects harbouring virus with Q148 with additional Q148 associated secondary mutations. The proportion of subjects with HIV RNA < 50 copies/mL at week 48 was 88/113 (78%) for no Q148 mutations, 19/31 (61%) for Q148 + 1 and 4/16 (25%) for Q148 + ≥ 2 secondary mutations (VO population, snapshot algorithm).
The response rate at week 48 was sustained with 116/183 (63% [95% CI: 56%, 70%]) subjects having HIV-1 RNA < 50 copies/mL (ITT-E, snapshot algorithm). Response was also sustained through week 48 in subjects harbouring virus with Q148 with additional Q148 associated secondary mutations. The proportion of subjects with HIV RNA < 50 copies/mL at week 48 was 88/113 (78%) for no Q148 mutations, 19/31 (61%) for Q148 + 1 and 4/16 (25%) for Q148 + ≥ 2 secondary mutations (VO population, snapshot algorithm). At Week 148 in the pooled SWORD-1 and SWORD-2 trials, 84% of subjects who received dolutegravir plus rilpivirine as of study start had plasma HIV-1 RNA < 50 copies/mL based on the Snapshot algorithm. In subjects who initially remained on their CAR and switched to dolutegravir plus rilpivirine at Week 52, 90% had plasma HIV-1 RNA < 50 copies/mL at Week 148 based on the Snapshot algorithm, which was comparable to the response rate (89%) observed at Week 100 (similar exposure duration) in subjects receiving dolutegravir plus rilpivirine as of study start.
At Week 148 in the pooled SWORD-1 and SWORD-2 trials, 84% of subjects who received dolutegravir plus rilpivirine as of study start had plasma HIV-1 RNA < 50 copies/mL based on the Snapshot algorithm. In subjects who initially remained on their CAR and switched to dolutegravir plus rilpivirine at Week 52, 90% had plasma HIV-1 RNA < 50 copies/mL at Week 148 based on the Snapshot algorithm, which was comparable to the response rate (89%) observed at Week 100 (similar exposure duration) in subjects receiving dolutegravir plus rilpivirine as of study start. There are no data available on the use of dolutegravir plus lamivudine as a two-drug regimen in paediatric patients.
There are no data available on the use of dolutegravir plus lamivudine as a two-drug regimen in paediatric patients.

