Notes
Distributed by Generic Health Pty Ltd
1 Name of Medicine
Tolvaptan.
2 Qualitative and Quantitative Composition
Tolvaptan Lupin tablets are for oral use and contain 15 mg, 30 mg, 45 mg, 60 mg or 90 mg of tolvaptan as the active ingredient.
List of excipients with known effects.
Lactose monohydrate.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Tolvaptan Lupin 15 mg.
Pink to light pink coloured, capsule shape, mottled tablet debossed with "FO5" on one side and "LU" on other side.
Tolvaptan Lupin 30 mg.
Pink to light pink coloured, round shape, flat faced bevelled mottled tablet debossed with "FO6" on one side and "LU" on other side.
Tolvaptan Lupin 45 mg.
Pink to light pink coloured, octagonal shape, mottled tablet debossed with "FO7" on one side and "LU" on other side.
Tolvaptan Lupin 60 mg.
Pink to light pink coloured, almond shaped, flat faced bevelled mottled tablet debossed with "FO8" on one side and "LU" on other side.
Tolvaptan Lupin 90 mg.
Pink to light pink coloured, capsule shape, biconvex mottled tablet debossed with "FO9" on one side and "LU" on other side.4.1 Therapeutic Indications
Tolvaptan Lupin is indicated to slow the progression of cyst development and renal insufficiency of autosomal dominant polycystic kidney disease (ADPKD) in adults with chronic kidney disease (CKD) stage 1 to 3 at initiation of treatment with evidence of rapidly progressing disease (see Section 5.1 Pharmacodynamic Properties).
4.2 Dose and Method of Administration
Tolvaptan Lupin treatment must be initiated and monitored under the supervision of physicians with expertise in managing ADPKD and a full understanding of the risks of tolvaptan therapy including hepatic toxicity and monitoring requirements (see Section 4.4 Special Warnings and Precautions for Use).
Dosage.
Tolvaptan Lupin is to be administered twice daily in split dose regimens of 45 mg + 15 mg, 60 mg + 30 mg or 90 mg + 30 mg. The morning dose is to be taken at least 30 minutes before the morning meal. The second daily dose can be taken with or without food. According to these split dose regimens the total daily doses are 60 mg, 90 mg or 120 mg.
Dose titration.
The initial dose is 60 mg tolvaptan per day as a split-dose regimen of 45 mg + 15 mg (45 mg taken upon waking and prior the morning meal and 15 mg taken 8 hours later). The initial dose is to be titrated upward to a split-dose regimen of 90 mg tolvaptan (60 mg + 30 mg) per day and then to a target split-dose regimen of 120 mg tolvaptan (90 mg + 30 mg) per day, if tolerated, with at least weekly intervals between titrations. Dose titration has to be performed cautiously to ensure that high doses are not poorly tolerated through overly rapid up-titration. Patients may down-titrate to lower doses based on tolerability. Patients have to be maintained on the highest tolerable tolvaptan dose.
The aim of dose titration is to block activity of vasopressin at the renal V2 receptor as completely and constantly as possible, while maintaining acceptable fluid balance, in order to achieve optimal effects on TKV progression or diminution of renal function decline. Measurements of urine osmolality are recommended to monitor the adequacy of vasopressin inhibition and, if possible, an absolute urine osmolality of less than 300 mOsm/kg should be maintained at all times (see Section 5.1 Pharmacodynamic Properties).
Also, monitoring of plasma osmolality or serum sodium (to calculate plasma osmolality) and/or body weight should be considered to monitor the risk of dehydration secondary to the aquaretic effects of tolvaptan in case of patient's insufficient water intake.
The safety and efficacy of tolvaptan in CKD stage 5 have not been adequately explored and therefore tolvaptan treatment should be discontinued if renal insufficiency progresses to CKD stage 5.
Therapy must be interrupted if the ability to drink or the accessibility to water is limited (see Section 4.4 Special Warnings and Precautions for Use). Tolvaptan must not be taken with grapefruit juice (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Patients must be instructed to drink sufficient amounts of water or other aqueous fluids (see Section 4.4 Special Warnings and Precautions for Use).
At the onset of symptoms or signs consistent with hepatic injury or if clinically significant abnormal ALT or AST increases are detected during treatment, Tolvaptan Lupin administration must be immediately interrupted and repeat tests including ALT, AST, BT and alkaline phosphatase (AP) must be obtained as soon as possible (ideally within 48 to 72 hours). Testing must continue at increased time frequency until symptoms/signs/laboratory abnormalities stabilise or resolve, at which point Tolvaptan Lupin may be cautiously reinitiated.
If the abnormal liver tests results are determined to be definitively related to tolvaptan therapy, restarting therapy is not recommended. However, if bilirubin, ALT and AST levels remained below the permanent discontinuation threshold, and if it is determined that tolvaptan would still be beneficial for the patient, tolvaptan therapy may be cautiously reinitiated with more frequent monitoring at the same or a lower dose.
In patients with a stable, low baseline AST or ALT, an increase above 2 times baseline, even if less than 2 times upper limit of normal, may indicate early liver injury. Such elevations may warrant treatment suspension and prompt (48 to 72 hours) re-evaluation of liver test trends prior to reinitiating therapy with more frequent monitoring.
Recommended guidelines for permanent discontinuation include:
ALT or AST > 8-times ULN;
ALT or AST > 5-times ULN for more than 2 weeks;
ALT or AST > 3-times ULN and (BT > 2-times ULN or International Normalised Ratio [INR] > 1.5);
ALT or AST > 3-times ULN with persistent symptoms of hepatic injury noted above.
Dose adjustment for patients taking strong CYP3A inhibitors.
In patients taking strong CYP3A inhibitors (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions), tolvaptan doses have to be reduced as follows (see Table 1).

Dose adjustment for patients taking moderate CYP3A inhibitors.
In patients taking moderate CYP3A inhibitors, tolvaptan doses have to be reduced as follows (see Table 2).
 Further reductions have to be considered if patients cannot tolerate the reduced tolvaptan doses.
Further reductions have to be considered if patients cannot tolerate the reduced tolvaptan doses.
Dosage adjustments for patients taking CYP3A inducers.
Concomitant use of tolvaptan with strong CYP3A inducers should be avoided (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Elderly population.
Increasing age has no effect on tolvaptan plasma concentrations. However, the safety and effectiveness of tolvaptan in ADPKD patients aged over 55 years has not yet been established.
Renal impairment.
Dose adjustment is not required in patients with renal impairment. No clinical trials in subjects with a creatinine clearance < 10 mL/min or in patients undergoing dialysis have been conducted. The risk of hepatic damage in patients with severely reduced renal function (i.e. eGFR < 20) may be increased; these patients should be carefully monitored for hepatic toxicity (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Hepatic impairment.
In patients with severe hepatic impairment, the benefits and risks of treatment with Tolvaptan Lupin must be evaluated carefully. Patients must be managed carefully and liver enzymes must be monitored regularly (see Section 4.4 Special Warnings and Precautions for Use).
Tolvaptan is contraindicated in patients with elevated liver enzymes and/or signs or symptoms of liver injury prior to initiation of treatment that meet the requirements for permanent discontinuation of tolvaptan (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
No dose adjustment is needed in patients with mild or moderate hepatic impairment (Child-Pugh classes A and B).
Paediatric population.
The safety and efficacy of tolvaptan in children and adolescents has not yet been established. No data are available. Tolvaptan is not recommended in the paediatric age group.
Method of administration.
For oral use.
Tablets must be swallowed without chewing and with a glass of water.4.3 Contraindications
Hypersensitivity to the active substance, benzazepine derivatives or to any of the excipients listed, see Section 6.1 List of Excipients.
Elevated liver enzymes and/or signs or symptoms of liver injury prior to initiation of treatment that meet the requirements for permanent discontinuation of tolvaptan (see Section 4.4 Special Warnings and Precautions for Use).
Volume depletion.
Anuria.
Hypernatremia.
Patients who cannot perceive or respond to thirst.
Pregnancy (see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy).
Breastfeeding (see Section 4.6 Fertility, Pregnancy and Lactation, Use in lactation).
4.4 Special Warnings and Precautions for Use
Idiosyncratic hepatic toxicity.
Tolvaptan has been associated with idiosyncratic elevations of blood alanine and aspartate aminotransferases (ALT and AST) with infrequent cases of concomitant elevations in bilirubin-total (BT).
In post-marketing experience with tolvaptan in ADPKD, acute liver failure requiring liver transplantation has been reported.
In a double-blind, placebo-controlled trial in patients with ADPKD, elevation (> 3 x upper limit of normal [ULN]) of ALT was observed in 4.4% (42/958) of patients on tolvaptan and 1.0% (5/484) of patients on placebo, while elevation (> 3 x ULN) of AST was observed in 3.1% (30/958) of patients on tolvaptan and 0.8% (4/484) patients on placebo. Two (2/957, 0.2%) of these tolvaptan treated-patients, as well as a third patient from an extension open label trial, exhibited increases in hepatic enzymes (> 3 x ULN) with concomitant elevations in BT (> 2 x ULN). The period of onset of hepatocellular injury (by ALT elevations > 3 x ULN) was within 3 to 14 months after initiating treatment and these increases were reversible, with ALT returning to < 3 x ULN within 1 to 4 months. While these concomitant elevations were reversible with prompt discontinuation of tolvaptan, they represent a potential for significant liver injury. Similar changes with other medicinal products have been associated with the potential to cause irreversible and potentially life-threatening liver injury.
Prescribing physicians must comply fully with the safety measures required below.
To mitigate the risk of significant and/or irreversible liver injury, blood testing for hepatic transaminases and bilirubin is required prior to initiation of Tolvaptan Lupin, continuing monthly for 18 months and at regular 3-monthly intervals thereafter. Concurrent monitoring for symptoms that may indicate liver injury (such as fatigue, anorexia, nausea, right upper abdominal discomfort, vomiting, fever, rash, pruritus, dark urine or jaundice) is recommended.
If a patient shows abnormal ALT, AST or BT levels prior to initiation of treatment which fulfil the criteria for permanent discontinuation (see below) tolvaptan treatment should not be initiated (see Section 4.3 Contraindications). In case of abnormal baseline levels below the limits for permanent discontinuation, pre-existing liver disease is likely, and treatment can only be initiated if the potential benefits of treatment outweigh the potential risks and liver function testing must continue at increased time frequency. The advice of a hepatologist is recommended.
During the first 18 months of treatment, Tolvaptan Lupin can only be supplied to patients whose physician has determined that liver function supports continued therapy.
At the onset of symptoms or signs consistent with hepatic injury or if clinically significant abnormal ALT or AST increases are detected during treatment, Tolvaptan Lupin administration must be immediately interrupted and repeat tests including ALT, AST, BT and alkaline phosphatase (AP) must be obtained as soon as possible (ideally within 48 to 72 hours). Testing must continue at increased time frequency until symptoms/signs/laboratory abnormalities stabilise or resolve, at which point Tolvaptan Lupin may be cautiously reinitiated.
If the abnormal liver tests results are determined to be definitively related to tolvaptan therapy, restarting therapy is not recommended. However, if bilirubin, ALT and AST levels remained below the permanent discontinuation threshold, and if it is determined that tolvaptan would still be beneficial for the patient, tolvaptan therapy may be cautiously reinitiated with more frequent monitoring at the same or a lower dose.
In patients with a stable, low baseline AST or ALT, an increase above 2 times baseline, even if less than 2 times upper limit of normal, may indicate early liver injury. Such elevations may warrant treatment suspension and prompt (48 to 72 hours) re-evaluation of liver test trends prior to reinitiating therapy with more frequent monitoring.
Recommended guidelines for permanent discontinuation include:
ALT or AST > 8-times ULN;
ALT or AST > 5-times ULN for more than 2 weeks;
ALT or AST > 3-times ULN and (BT > 2-times ULN or International Normalized Ratio [INR] > 1.5);
ALT or AST > 3-times ULN with persistent symptoms of hepatic injury noted above.
Because of the risk of liver injury, prescriber education and certification on the risk of liver injury and the importance of regular liver function monitoring is mandatory. These are available through the Tolvaptan Portal on the sponsor website.
Further information is available at www.generichealth.com.au or by telephone at 03 9809 7900.
Access to water.
Tolvaptan may cause adverse reactions related to water loss such as thirst, polyuria, nocturia, and pollakiuria (see Section 4.8 Adverse Effects (Undesirable Effects)). Therefore, patients must have access to water (or other aqueous fluids) and be able to drink sufficient amounts of these fluids (see Section 4.2 Dose and Method of Administration). Patients have to be instructed to drink water or other aqueous fluids at the first sign of thirst in order to avoid excessive thirst or dehydration.
Additionally, patients have to drink 1 to 2 glasses of fluid before bedtime regardless of perceived thirst and replenish fluids overnight with each episode of nocturia.
Dehydration.
Volume status must be monitored in patients taking tolvaptan because treatment with tolvaptan may result in severe dehydration which constitutes a risk factor for renal dysfunction. If dehydration becomes evident, take appropriate action which may include the need to interrupt or reduce the dose of tolvaptan and increase fluid intake. Special care must be taken in patients having diseases that impair appropriate fluid intake or who are at an increased risk of water loss e.g. in case of vomiting or diarrhoea. In such circumstances patients should be advised to cease tolvaptan treatment and seek urgent medical assistance.
Patients exposed to prolonged heat, humidity, exercise and intercurrent illness will be at greater risk of dehydration.
Urinary outflow obstruction.
Urinary output must be secured. Patients with partial obstruction of urinary outflow, for example patients with prostatic hypertrophy or impairment of micturition, have an increased risk of developing acute retention.
Fluid and electrolyte balance.
Fluid and electrolyte status must be monitored in all patients. Administration of tolvaptan induces copious aquaresis and may cause dehydration and increases in serum sodium (see Section 4.8 Adverse Effects (Undesirable Effects)) and is contraindicated in hypernatraemic patients (see Section 4.3 Contraindications). Therefore, serum creatinine, electrolytes and symptoms of electrolyte imbalances (e.g. dizziness, fainting, palpitations, confusion, weakness, gait instability, hyper-reflexia, seizures, coma) have to be assessed prior to and after starting tolvaptan to monitor for dehydration.
During long-term treatment electrolytes have to be monitored at least every three months.
Serum sodium abnormalities.
Pretreatment sodium abnormalities (hyponatraemia or hypernatraemia) must be corrected prior to initiation with tolvaptan therapy.
Hyperkalemia.
Treatment with tolvaptan is associated with an acute reduction of the extracellular fluid volume which could results in increased serum potassium. Serum potassium levels should be monitored carefully after initiation of tolvaptan, especially in those who are receiving drugs known to increase serum potassium levels.
Anaphylaxis.
In post-marketing experience, anaphylaxis (including anaphylactic shock and rash generalised) has been reported very rarely following administration of tolvaptan. This type of reaction occurred after the first administration of tolvaptan. If an anaphylactic reaction or other serious allergic reactions occur, administration of tolvaptan must be discontinued immediately and appropriate therapy initiated. Since hypersensitivity is a contraindication (see Section 4.3 Contraindications) treatment must never be restarted after an anaphylactic reaction or other serious allergic reactions.
Lactose.
Tolvaptan Lupin contains lactose as an excipient. Patients with rare hereditary problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose malabsorption should not take this medicine.
Diabetes mellitus.
Diabetic patients with an elevated glucose concentration (e.g. in excess of 300 mg/dL) may present with pseudohyponatraemia. This condition must be excluded prior and during treatment with tolvaptan.
Tolvaptan may cause hyperglycaemia (see Section 4.8 Adverse Effects (Undesirable Effects)). Therefore, diabetic patients treated with tolvaptan must be managed cautiously. In particular this applies to patients with inadequately controlled type II diabetes.
Uric acid increases.
Decreased uric acid clearance by the kidney is a known effect of tolvaptan. In a double-blind, placebo-controlled trial of patients with ADPKD, potentially clinically significant increased uric acid (greater than 10 mg/dL) was reported at a higher rate in tolvaptan-patients (6.2%) compared to placebo-treated patients (1.7%). Adverse reactions of gout were reported more frequently in tolvaptan-treated patients (28/961, 2.9%) than in patients receiving placebo (7/483, 1.4%). In addition, increased use of allopurinol and other medicinal products used to manage gout were observed in the double-blind, placebo-controlled trial. Effects on serum uric acid are attributable to the reversible renal hemodynamic changes that occur in response to tolvaptan effects on urine osmolality and may be clinically relevant. However, events of increased uric acid and/or gout were not serious and did not cause discontinuation of therapy in the double-blind, placebo-controlled trial. Uric acid concentrations are to be evaluated prior to initiation of Tolvaptan Lupin therapy, and as indicated during treatment based on symptoms.
Effect of tolvaptan on glomerular filtration rate (GFR).
A reversible reduction in GFR has been observed in ADPKD trials at the initiation of tolvaptan treatment.
Use in the elderly.
Increasing age has no effect on tolvaptan plasma concentrations. However, the safety and effectiveness of tolvaptan in ADPKD patients aged over 55 years has not yet been established.
Paediatric use.
No data are available. Tolvaptan is not recommended in the paediatric age group.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other medicinal products on the pharmacokinetics of tolvaptan.
CYP3A inhibitors.
Concomitant use of medicinal products that are moderate (e.g. amprenavir, aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil) or strong (e.g. itraconazole, ketoconazole, ritonavir, clarithromycin, saquinavir) CYP3A inhibitors increase tolvaptan exposure. Co-administration of tolvaptan and ketoconazole resulted in a 440% increase in area under time-concentration curve (AUC) and 248% increase in maximum observed plasma concentration (Cmax) for tolvaptan.
Co-administration of tolvaptan with 240 mL grapefruit juice, a moderate to strong CYP3A inhibitor, produced a doubling of peak tolvaptan concentrations (Cmax).
Dose reduction of tolvaptan is recommended for patients while taking moderate or strong CYP3A inhibitors (see Section 4.2 Dose and Method of Administration). Patients taking moderate or strong CYP3A inhibitors must be managed cautiously, in particular if the inhibitors are taken more frequently than once a day.
CYP3A inducers.
Concomitant use of medicinal products that are potent CYP3A inducers (e.g. rifampicin) will decrease tolvaptan exposure and efficacy. Co-administration of tolvaptan with rifampicin reduces Cmax and AUC for tolvaptan by about 85%. Therefore, concomitant administration of tolvaptan with potent CYP3A inducers (e.g. rifampicin, rifabutin, rifapentin, phenytoin, carbamazepine, and St. John's wort) is to be avoided.
P-gp inhibitors.
Reduction in the dose of tolvaptan may be required in patients concomitantly treated with P-glycoprotein (P-gp) inhibitors, such as cyclosporine and quinidine, based on clinical response (see Section 4.2 Dose and Method of Administration). If P-gp inhibitors also act as strong CYP3A inhibitors (e.g. ketoconazole, clarithromycin, ritonavir, saquinavir), substantial dose reduction of tolvaptan is required (see Section 4.2 Dose and Method of Administration).
Co-administration with medicinal products that increase serum sodium concentration.
There is no experience from controlled clinical trials with concomitant use of tolvaptan and hypertonic saline, oral sodium formulations, and medicinal products that increase serum sodium concentration. Medicinal products with high sodium content such as effervescent analgesic preparations and certain sodium containing treatments for dyspepsia may also increase serum sodium concentration. Concomitant use of tolvaptan with medicinal products that increase serum sodium concentration may result in a higher risk for developing hypernatraemia (see Section 4.4 Special Warnings and Precautions for Use) and is therefore not recommended.
Diuretics.
Tolvaptan has not been extensively studied in ADPKD in combination with diuretics. While there does not appear to be a synergistic or additive effect of concomitant use of tolvaptan with loop and thiazide diuretics, each class of agent has the potential to lead to severe dehydration, which constitutes a risk factor for renal dysfunction. If dehydration or renal dysfunction becomes evident, appropriate action must be taken which may include the need to interrupt or reduce doses of tolvaptan and/or diuretics and increased fluid intake. Other potential causes of renal dysfunction or dehydration must be evaluated and addressed.
Effect of tolvaptan on the pharmacokinetics of other products.
CYP3A substrates.
In healthy subjects, tolvaptan, a CYP3A substrate, had no effect on the plasma concentrations of some other CYP3A substrates (e.g. warfarin or amiodarone). Tolvaptan increased plasma levels of lovastatin by 1.3 to 1.5-fold. Even though this increase has no clinical relevance, it indicates tolvaptan can potentially increase exposure to CYP3A4 substrates.
Transporter substrates.
In vitro studies indicate that tolvaptan is a substrate and competitive inhibitor of P-gp. In vitro studies indicate that tolvaptan or its oxobutyric metabolite may have the potential to inhibit OATP1B1, OATP1B3, OAT3, BCRP and OCT1 transporters.
Steady state digoxin concentrations were increased (1.3-fold in maximum observed plasma concentration [Cmax] and 1.2-fold in area under the plasma concentration-time curve over the dosing interval [AUCτ]) when co-administered with multiple once daily 60 mg doses of tolvaptan. Patients receiving digoxin or other narrow therapeutic P-gp substrates (e.g. dabigatran) must therefore be managed cautiously and evaluated for excessive effects when treated with tolvaptan.
Statins commonly used in the tolvaptan phase 3 pivotal trial (e.g. rosuvastatin and pitavastatin) are OATP1B1 or OATP1B3 substrates, however no difference in AE profile was observed during the phase 3 pivotal trial for tolvaptan in ADPKD.
If OATP1B1 and OATP1B3 substrates (e.g. statins such as rosuvastatin and pitavastatin), OAT3 substrates (e.g. methotrexate, ciprofloxacin), BCRP substrates (e.g. sulfasalazine) or OCT1 substrates (e.g. metformin) are co-administered with tolvaptan, patients must be managed cautiously and evaluated for excessive effects of these medications.
Diuretics or non-diuretic anti-hypertensive drug(s).
Standing blood pressure was not routinely measured in ADPKD trials, therefore a risk of orthostatic/postural hypotension due to a pharmacodynamic interaction with tolvaptan cannot be excluded.
Co-administration with vasopressin analogues.
In addition to its renal aquaretic effect, tolvaptan is capable of blocking vascular vasopressin V2 receptors involved in the release of coagulation factors (e.g. von Willebrand factor) from endothelial cells. Therefore, the effect of vasopressin analogues such as desmopressin may be attenuated in patients using such analogues to prevent or control bleeding when co-administered with tolvaptan. It is not recommended to administer Tolvaptan Lupin with vasopressin analogues.
Smoking and alcohol.
Data related to smoking or alcohol history in ADPKD trials are too limited to determine possible interactions of smoking or alcohol with efficacy and safety of ADPKD treatment with tolvaptan.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Fertility was unaffected in male and female rats given tolvaptan at oral doses up to 1000 mg/kg/day (yielding 1.9-times in males and 5.1-times in females the AUC in patients at the maximum recommended human dose [MRHD] of 120 mg per day). However, oestrus cycles were altered in rats at oral doses ≥ 300 mg/kg/day (3.8-times the clinical AUC at the MRHD).
(Category D)
Tolvaptan and/or its metabolites were shown to cross the placenta in rats and rabbits.
In rats treated with tolvaptan during organogenesis, reduced foetal weights and delayed foetal ossification occurred at an oral dose of 1000 mg/kg/day (yielding 17-times the clinical AUC at the MRHD).
Teratogenicity was noted in rabbits given 1000 mg/kg/day (7.5 times the exposure from the 120 mg/day human dose on an AUC basis). The effects were increased rates of embryo-foetal death, foetal microphthalmia, open eyelids, cleft palate, brachymelia and skeletal malformations. These adverse effects on embryo-foetal development were observed in conjunction with maternal toxicity (reduced maternal body weight gain and food consumption) although a direct effect of the drug cannot be excluded.
No teratogenic effects were seen in rabbits at 300 mg/kg/day (about 1.25 to 2.65 times the exposure in humans at the 120 mg/day dose, based on AUC). However, increasing the dose from 300 mg/kg/day to 1000 mg/kg/day increased abortions from 1/17 to 5/18 animals.
There are no adequate data from the use of tolvaptan in pregnant women. The potential risk for humans is unknown.
Women of childbearing potential must use adequate contraceptive measures during Tolvaptan Lupin use. Tolvaptan Lupin must not be used during pregnancy (see Section 4.3 Contraindications).
It is unknown whether tolvaptan is excreted in human breast milk. Studies in rats have shown excretion of tolvaptan in milk.
The potential risk for humans is unknown. Tolvaptan Lupin is contraindicated during breastfeeding (see Section 4.3 Contraindications).4.7 Effects on Ability to Drive and Use Machines
Tolvaptan Lupin has minor influence on the ability to drive or use machines. However, when driving vehicles or using machines it has to be taken into account that occasionally dizziness, asthenia or fatigue may occur.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
The pharmacodynamically predictable and most commonly reported adverse reactions are thirst, polyuria, nocturia and pollakiuria occurring in approximately 55%, 38%, 29% and 23% of patients, respectively. Furthermore, tolvaptan has been associated with idiosyncratic elevations of blood alanine and aspartate aminotransferases (ALT and AST) with infrequent cases of concomitant elevations in bilirubin-total (BT).
Tabulated list of adverse reactions.
The adverse reaction profile of tolvaptan in the ADPKD indication is based on the results of two clinical trials: 156-04-251 of 1,444 treated patients (961 patients treated with tolvaptan, 483 treated with placebo) and 156-13-210 of 1,366 patients (681 patients treated with tolvaptan, 685 treated with placebo), and is consistent with the pharmacology of the active substance.
Adverse reactions associated with tolvaptan obtained from ADPKD clinical studies are tabulated in Table 3 and Table 4.
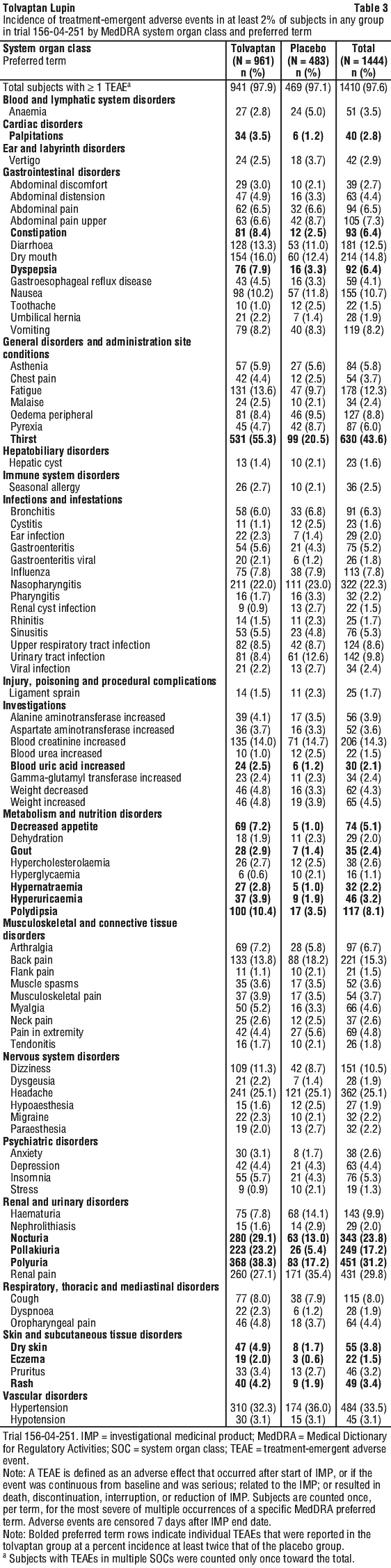
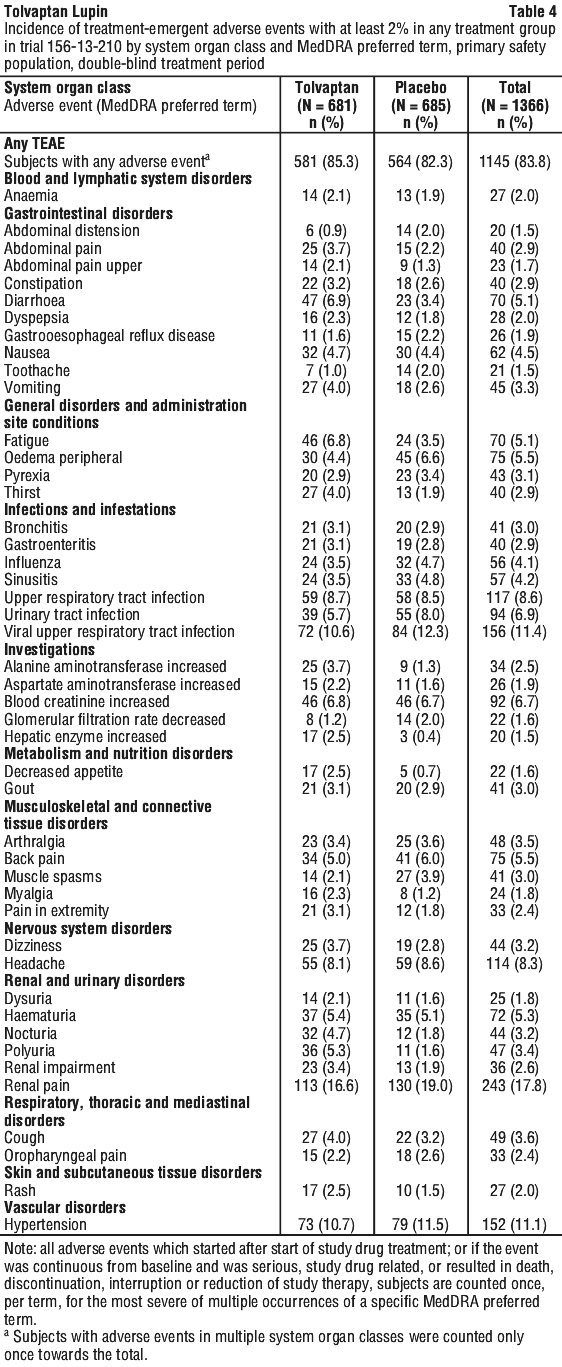
Description of selected adverse reactions.
To mitigate the risk of significant or irreversible liver injury, blood testing for hepatic transaminases is required prior to initiation of Tolvaptan Lupin treatment, continuing monthly for 18 months and at regular 3-monthly intervals thereafter (see Section 4.4 Special Warnings and Precautions for Use).
The most frequent adverse reactions are related to water loss. It is therefore of greatest importance that patients have access to water and are able to drink sufficient amounts of fluids.
The volume status of patients taking tolvaptan must be monitored to prevent dehydration (see Section 4.4 Special Warnings and Precautions for Use).
Post-marketing reports.
The following adverse reactions have been reported during post-marketing surveillance of tolvaptan. The exact incidence of spontaneously reported adverse reactions is unknown: anaphylaxis; generalised rash.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Single oral doses up to 480 mg (4 times the maximum recommended daily dose) and multiple doses up to 300 mg once daily for 5 days have been well tolerated in trials in healthy subjects. There is no specific antidote for tolvaptan intoxication. The signs and symptoms of an acute overdose can be anticipated to be those of excessive pharmacologic effect: a rise in serum sodium concentration, polyuria, thirst and dehydration/hypovolemia.
No mortality was observed in rats or dogs following single oral doses of 2000 mg/kg (maximum feasible dose). A single oral dose of 2000 mg/kg was lethal in mice and symptoms of toxicity in affected mice included decreased locomotor activity, staggering gait, tremor and hypothermia.
In patients with suspected tolvaptan overdose, assessment of vital signs, electrolyte concentrations, ECG and fluid status is recommended. Appropriate replacement of water and/or electrolytes must continue until aquaresis abates. Dialysis may not be effective in removing tolvaptan because of its high binding affinity for human plasma protein (> 98%).
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Tolvaptan is a vasopressin antagonist: ATC code C03XA01.
Mechanism of action.
Tolvaptan is a vasopressin antagonist that specifically blocks the binding of arginine vasopressin (AVP) at the V2 receptors of the distal portions of the nephron. Tolvaptan affinity for the human V2 receptor is 1.8 times that of native AVP.
Pharmacodynamic effects.
The pharmacodynamic effects of tolvaptan have been determined in healthy subjects and subjects with autosomal dominant polycystic kidney disease (ADPKD) across chronic kidney disease (CKD) stages 1 to 4. Effects on free water clearance and urine volume are evident across all CKD stages with smaller absolute effects observed at later stages, consistent with the declining number of fully functioning nephrons. Acute reductions in mean total kidney volume were also observed following 3 weeks of therapy in all CKD stages, ranging from -4.6% for CKD stage 1 to -1.9% for CKD stage 4.
With the recommended split-dose regimens in patients with autosomal dominant polycystic kidney disease (ADPKD), tolvaptan inhibits vasopressin from binding to the V2 receptor in the kidney for the entire day, as indicated by increased urine output and decreased urine osmolality to below 300 mOsm/kg. Higher doses, e.g. 90 + 30 mg/day, reduced urine osmolality to below 300 mOsm/kg in a greater proportion of patients than the lower doses.
In healthy subjects or patients with CKD stage 1 to 4 receiving a single dose of tolvaptan, the onset of the aquaretic effects occurs within 1 to 2 hours post-dose and peaks between 4 and 8 hours post-dose. Higher doses of tolvaptan do not increase the peak effect in urine excretion rate but sustain the effect for a longer period of time. The offset of tolvaptan action is rapid with urine excretion rate returning to baseline within 24 hours following a 90 mg dose.
Increases in daily urine output in response to tolvaptan treatment are positively correlated with baseline renal function. Following a 90 + 30 mg split-dose regimen in patients with CKD stage 1 or 2, the change in mean daily urine volume was about 4 L for a mean total daily volume of about 7 L. In stage 4 patients, the mean change in daily urine volume was about 2 L for a total daily urine volume of about 5 L.
Clinical trials.
The primary focus of the clinical program for development of tolvaptan tablets for the treatment of ADPKD is a single pivotal, multinational, phase 3, randomised, placebo-controlled trial in which the long-term safety and efficacy of oral split dose tolvaptan regimens (titrated between 60 mg/day and 120 mg/day) were compared with placebo in 1,445 adult subjects with ADPKD. In total, 14 clinical trials involving tolvaptan have been completed worldwide in support of the ADPKD indication, including 8 trials in the US, 1 in the Netherlands, 3 in Japan, 1 in Korea, and the multinational phase 3 pivotal trial.
In the pivotal double-blind, 36-month, placebo-controlled, multi-centre trial (TEMPO 3:4), a total of 1,445 adult patients (age 18 to 50 years) with early, rapidly-progressing ADPKD (meeting modified Ravine criteria, total kidney volume (TKV) ≥ 750 cc, estimated creatinine clearance ≥ 60 mL/min) were randomised 2:1 to treatment with tolvaptan or placebo, respectively. A total of 1,445 patients were treated for up to 3 years, then followed for 14 to 42 days after treatment withdrawal. Randomisation was stratified based on several predictors of more rapid progression, namely baseline hypertension status, kidney volume and renal function. Other known predictors of risk of rapid progression, not used in the stratification, include a family history of ADPKD with early end-stage renal disease (ESRD) or high bilateral cyst number. All patients remained on standard concomitant medications. Concomitant use of potent cytochrome P450 CYP3A4 inhibitors was avoided. Investigational drug was up-titrated from a daily, oral split dose of 45 mg on waking plus 15 mg 9 hours later (45 mg/15 mg) weekly, as tolerated to 60 mg/30 mg then 90 mg/30 mg. Patients were to maintain the highest tolerated dose for 3 years, but could interrupt, decrease and/or increase as clinical circumstances warranted within the range of titrated doses. All patients were encouraged to drink adequate water to avoid thirst or dehydration and at night before retiring. Patients were evaluated at screening, baseline, during weekly titration steps and at intervals of at least 4 months for outcome, laboratory and safety assessments. At baseline and yearly visits, MRI-TKV and pharmacokinetic assessments were also performed. Patients who completed the study terminated treatment at 3 years, and were followed for a further period of 2 to 6 weeks to assess off-drug effects.
Tolvaptan (N=961) and placebo (N=484) groups were well matched and representative of regional populations with an average age of 39 years, with 52% being male, 84% Caucasian, 13% Asian and 3% other races. At baseline 79% had hypertension, average estimated glomerular filtration rate (eGFR) was 82 mL/min/1.73 m2 (Chronic Kidney Disease Epidemiology formula (CKD-EPI)) and mean TKV was 1692 cc (height adjusted 972 cc/m). Using CKD-EPI eGFR, tolvaptan and placebo patients were distributed across Kidney Disease Outcomes Quality Initiative chronic kidney disease (KDOQI-CKD) stages 1 (34% and 36%), 2 (49% and 46%) and 3 (17% and 17%), respectively. Within the study population receiving placebo, stratification factors successfully predicted more rapid progression in those who had larger kidneys, lower eGFR, or hypertension at baseline. No anti-hypertensive medications were being taken by 23% of patients; in the remaining 77% of patients, 71% were taking agents acting on the renin-angiotensin system, 20% calcium channel blockers and, 18% beta blockers. Analgesics were used in 10% of patients for unspecified indications while an additional 5% of patients used these for kidney pain. Dietary restrictions for sodium, protein or caffeine were prescribed in 31%, 17% and 34% of patients, respectively.
The primary endpoint (TKV slope) was the intergroup difference for rate of change of TKV normalised as a percentage. TKV increased in the tolvaptan group at a rate of 2.80% per year (95% confidence interval [CI], 2.5% to 3.1%) compared with 5.51% per year (95% CI, 5.1% to 6.0%) for placebo representing a 49.2% reduction in growth rate averaged over 3 years (p < 0.0001).
Pre-specified secondary end-points were tested sequentially. The key secondary composite endpoint (ADPKD progression) was time to multiple clinical progression events of: 1) worsening kidney function (defined as a persistent [reproduced over at least 2 weeks] 25% reduction in reciprocal serum creatinine during treatment (from end of titration to last on-drug visit); 2) medically significant kidney pain (defined as requiring prescribed leave, last-resort analgesics, narcotic and anti-nociceptive, radiologic or surgical interventions); 3) worsening hypertension (defined as a persistent increase in blood pressure category or an increased anti-hypertensive prescription); 4) worsening albuminuria (defined as a persistent [reproduced at 2 of 3 successive assessments] increase in albumin/creatinine ratio category).
The relative rate of ADPKD-related events was decreased by 13.5% in tolvaptan-treated patients, (44 vs. 50 events; hazard ratio, 0.87; 95% CI, 0.78 to 0.97; p=0.0095).
The result of the key secondary composite endpoint is primarily attributed to effects on worsening kidney function and medically significant kidney pain. The renal function events were 61.4% less likely for tolvaptan compared with placebo (2 events per 100 person-years of follow-up in the tolvaptan group vs. 5 in the placebo group; hazard ratio, 0.39; 95% CI, 0.26 to 0.57; nominal p < 0.0001, see Figure 1), while renal pain events were 35.8% less likely in tolvaptan-treated patients (5 events per 100 person-years of follow-up in the tolvaptan group vs. 7 in the placebo group; hazard ratio, 0.64; 95% CI, 0.47 to 0.89; nominal p = 0.007, see Figure 1). In contrast, there was no effect of tolvaptan on either progression of hypertension or albuminuria.
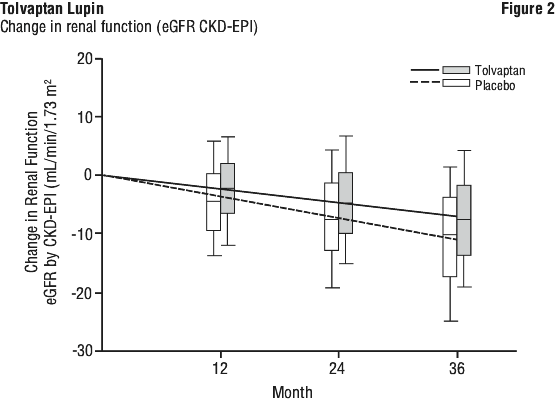
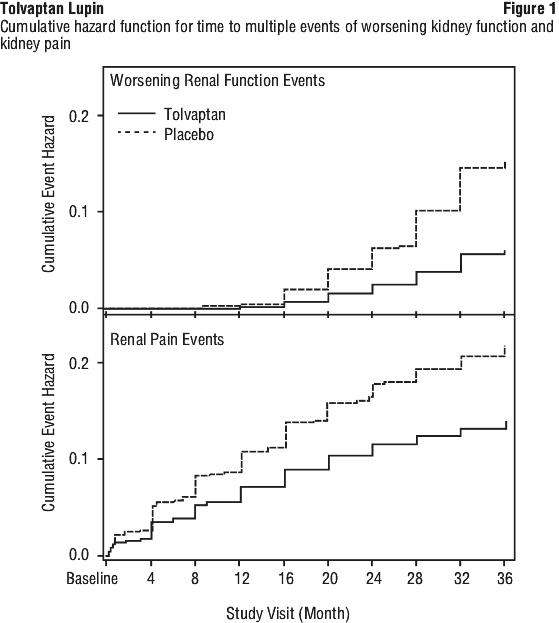 The next sequentially ordered secondary endpoint of slope of kidney function decline was assessed as change in estimated glomerular filtration rate (eGFR-CKD EPI) during treatment (from end of titration to last on-drug visit). The tolvaptan-treated patients had a 26.4% reduction in the rate of renal function decline compared with placebo (-2.7 mL/min/1.73 m2 versus -3.7 mL/min/1.73 m2, p < 0.0001, Figure 2). Figure 2 represents slope of renal function for tolvaptan (solid) and placebo (dashed) change from end of titration baseline (i.e. end of week 3). Box plots derived from mixed effect model repeat measurement (MMRM) analyses to each indicated 12-month visit with 5th, 25th, mean, 75th and 95th percentiles of change from end of titration for tolvaptan (grey) and placebo (white) groups.
The next sequentially ordered secondary endpoint of slope of kidney function decline was assessed as change in estimated glomerular filtration rate (eGFR-CKD EPI) during treatment (from end of titration to last on-drug visit). The tolvaptan-treated patients had a 26.4% reduction in the rate of renal function decline compared with placebo (-2.7 mL/min/1.73 m2 versus -3.7 mL/min/1.73 m2, p < 0.0001, Figure 2). Figure 2 represents slope of renal function for tolvaptan (solid) and placebo (dashed) change from end of titration baseline (i.e. end of week 3). Box plots derived from mixed effect model repeat measurement (MMRM) analyses to each indicated 12-month visit with 5th, 25th, mean, 75th and 95th percentiles of change from end of titration for tolvaptan (grey) and placebo (white) groups.
 Subgroup analysis of all endpoints above (change in TKV, key composite [including time to worsening of kidney function and time to medically significant kidney pain] and change in slope of decline in renal function) demonstrated consistent efficacy (directional) in all pre-specified subgroups, including those stratified by age, gender, race, geographical location, baseline hypertension, baseline eGFR and baseline TKV.
Subgroup analysis of all endpoints above (change in TKV, key composite [including time to worsening of kidney function and time to medically significant kidney pain] and change in slope of decline in renal function) demonstrated consistent efficacy (directional) in all pre-specified subgroups, including those stratified by age, gender, race, geographical location, baseline hypertension, baseline eGFR and baseline TKV.
Patients were not genotyped to separate into ADPKD type 1 and 2 and it is not known whether tolvaptan has comparable efficacy in these subgroups.
The Phase 3, multi-centre, international, randomised-withdrawal, placebo-controlled, double-blind trial 156-13-210 (REPRISE) compared the efficacy and safety of tolvaptan (45 to 120 mg/day) to placebo in later stage ADPKD (CKD stage 2 to 4). A total of 1,370 patients (age 18 to 65) were randomised to either tolvaptan (n = 683) or placebo (n = 687) and were treated for a period of 12 months. Approximately 5%, 75% and 20% had an eGFR 60 mL/min/1.73 m2 or greater (CKD stage 2), or less than 60 mL/min/1.73 m2 and greater than 30 mL/min/1.73 m2 (CKD stage 3) or less than 30 mL/min/1.73 m2 but greater than 15 mL/min/1.73 m2 (CKD stage 4), respectively.
The primary endpoint of the trial was the change in estimated glomerular filtration rate (eGFR) from pre-treatment baseline levels to post-treatment assessment. In patients treated with tolvaptan the reduction in eGFR was significantly less than in patients treated with placebo (p < 0.0001).
The treatment difference in eGFR change observed in this trial is 1.27 mL/min/1.73 m2, representing a 35% reduction in the loss of kidney function compared to placebo over the course of one year (see Table 5).
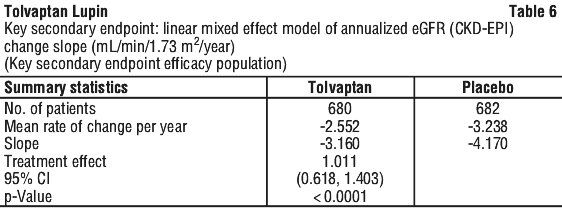
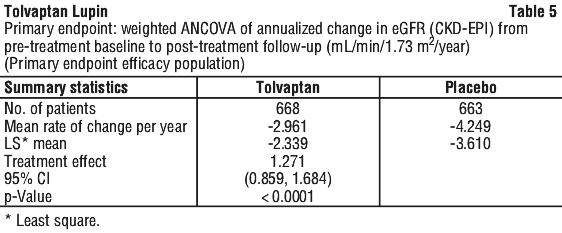 The key secondary endpoint was a comparison of the efficacy of tolvaptan treatment versus placebo in reducing the decline of annualised eGFR slope across all measured time points in the trial. These data also showed statistically significant benefit from tolvaptan vs. placebo (p < 0.0001) (see Table 6).
The key secondary endpoint was a comparison of the efficacy of tolvaptan treatment versus placebo in reducing the decline of annualised eGFR slope across all measured time points in the trial. These data also showed statistically significant benefit from tolvaptan vs. placebo (p < 0.0001) (see Table 6).
 Pre-specified subgroup analyses of the primary endpoint showed a beneficial effect of tolvaptan across subgroups that were defined according to sex, baseline estimated GFR, stage of chronic kidney disease (except for stage 2) and geographic region, as well as in the subgroups of patients who were 55 years of age or younger and patients who were white.
Pre-specified subgroup analyses of the primary endpoint showed a beneficial effect of tolvaptan across subgroups that were defined according to sex, baseline estimated GFR, stage of chronic kidney disease (except for stage 2) and geographic region, as well as in the subgroups of patients who were 55 years of age or younger and patients who were white.
The pre-specified subgroup analysis was inconclusive in the smaller sample size subgroups of patients who were non-white, or who were older than 55 years of age, a subgroup which also had a notably slower rate of eGFR decline. The prespecified analysis was not significant for patients who had chronic kidney disease of stage 2 due to the limited sample size in REPRISE trial.
Safety results were consistent with the known safety profile based on previous tolvaptan clinical trials.
In a long-term, open-label, multi-centre dose ranging phase 2 trial (TEMPO 2:4) in 46 adult patients (age 18 to 50 years) with ADPKD (meeting modified Ravine criteria and an estimated GFR ≥ 30 mL/min) patients were titrated in weekly intervals for the first 4 weeks among split doses of 30 mg/15 mg (down-titration to 15 mg/15 mg allowed for tolerability) to 90 mg/30 mg, then maintained at their maximal tolerated dose (MTD) through Month 2 (Titration Period). Following a determination of final long-term dosing regimens based on objective criteria (trough spot urine osmolality < 300 mOsm/kg, median tolerability) using data from the Titration Period, patients were randomly allocated to a fixed high dose regimen (60 mg/30 mg) and fixed low dose regimen (45 mg/15 mg) for up to Month 36 (Fixed-dose Period). All patients discontinued treatment at the end of this phase of the trial and after a period of 4 to 6 months had an option to enter an Extension Period of treatment for an additional 12 months.
All 46 of the patients who entered the Titration Period were treated in the Fixed-dose Period, including 22 patients in the tolvaptan 45 mg/15 mg group and 24 patients in the tolvaptan 60 mg/30 mg group. Over the course of 36 to 48 months of open-label tolvaptan treatment, 39/46 patients (84.8%) completed the 36-month portion of the trial and 35/35 patients (100%) completed the 12-month Extension. At Month 36, TKV increased from baseline in both groups, with a higher mean (SD) percent increase seen in the tolvaptan 45 mg/15 mg group (9.86% [11.81%]) compared with the tolvaptan 60 mg/30 mg group 5.06% [9.77%]; nominal p = 0.0553). The mean (SD) annual rate of change from pre-dose baseline in creatinine clearance estimate by Cockcroft-Gault equation (eCrCLCG) was -2.17 (14.45) mL/min/year in the tolvaptan 45 mg/15 mg group and -0.450 (4.86) mL/min/year in the tolvaptan 60 mg/30 mg group. Upon withdrawal from treatment, TKV increased at higher rates than while on treatment. Upon resumption of treatment, in the extension phase rates of TKV growth again slowed (see Figure 3). These results support that patients treated with higher doses of tolvaptan have a greater treatment effect and that continued treatment is required to maintain benefits and provided support for the dosing recommendations in the pivotal trial discussed above.
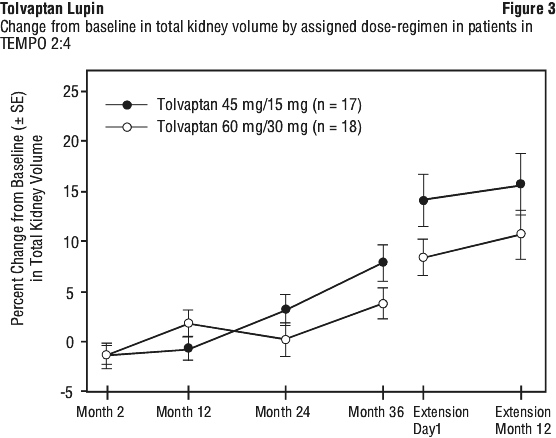 Data are not currently available to show whether long-term therapy with tolvaptan continues to slow the rate of renal function decline and affect clinical outcomes of ADPKD, including delay in the onset of end-stage renal disease.
Data are not currently available to show whether long-term therapy with tolvaptan continues to slow the rate of renal function decline and affect clinical outcomes of ADPKD, including delay in the onset of end-stage renal disease.
5.2 Pharmacokinetic Properties
Absorption and distribution.
After oral administration, tolvaptan is rapidly absorbed with peak plasma concentrations occurring about 2 hours after dosing. The absolute bioavailability of tolvaptan is about 56%.
Co-administration of tolvaptan with a high-fat meal increased peak concentrations of tolvaptan up to 2-fold but left AUC unchanged. Even though the clinical relevance of this finding is not known, to minimise the unnecessary risk of increasing the maximal exposure the morning dose should be taken under fasted conditions (see Section 4.2 Dose and Method of Administration).
Following single oral doses of 300 mg, peak plasma concentrations appear to plateau, possibly due to saturation of absorption. Tolvaptan binds reversibly (98%) to plasma proteins.
Metabolism.
Tolvaptan is extensively metabolised in the liver almost exclusively by CYP3A. Tolvaptan is a weak CYP3A4 substrate and does not appear to have any inhibitory activity.
In vitro studies indicated that tolvaptan has no inhibitory activity for CYP3A. Fourteen metabolites have been identified in plasma, urine and faeces; all but one were also metabolised by CYP3A. Only the oxobutyric acid metabolite is present at greater than 10% of total plasma radioactivity; all others are present at lower concentrations than tolvaptan.
Tolvaptan metabolites have little to no contribution to the pharmacological effect of tolvaptan; all metabolites have no or weak antagonist activity for human V2 receptors when compared with tolvaptan.
Excretion.
The terminal elimination half-life is about 8 hours and steady-state concentrations of tolvaptan are obtained after the first dose.
Less than 1% of intact active substance is excreted unchanged in the urine. Radio labelled tolvaptan experiments showed that 40% of the radioactivity was recovered in the urine and 59% was recovered in the faeces, where unchanged tolvaptan accounted for 32% of radioactivity. Tolvaptan is only a minor component in plasma (3%).
Linearity.
Following single oral doses, Cmax values show less than dose proportional increases from 30 mg to 240 mg and then a plateau at doses from 240 mg to 480 mg, AUC increases linearly.
Following multiple once daily dosing of 300 mg, tolvaptan exposure was only increased 6.4-fold when compared to a 30 mg dose. Based on calculations from 4 different studies in split-dose regimens of 30 mg, 60 mg and 120 mg/day in ADPKD patients, tolvaptan exposure (AUC) increases approximately linearly.
Pharmacokinetics in special populations.
Elderly.
Clearance of tolvaptan is not significantly affected by age.
Hepatic dysfunction.
The effect of mildly or moderately impaired hepatic function (Child-Pugh classes A and B) on the pharmacokinetics of tolvaptan was investigated in 87 patients with liver disease of various origins. For single doses of 10 mg to 60 mg, Cmax and AUC∞ increase linearly with dose. Although mean Cmax values for 30 mg and 60 mg doses do not appear to be much higher compared to healthy subjects, 1.07 to 1.65-fold, AUC values are 1.56 to 2.75-fold higher. Following multiple dosing (QD, 13 days) tolvaptan concentrations also appear to accumulate 1.7 to 1.6-fold. Clearance following a single dose is about half that of healthy subjects and following multiple dosing is about a third that of healthy subjects. Very limited information is available in patients with severe hepatic impairment (Child-Pugh class C).
In a population pharmacokinetic analysis in patients with hepatic oedema, AUC of tolvaptan in severely (Child-Pugh class C) and mildly or moderately (Child-Pugh classes A and B) hepatic impaired patients were 3.1 and 2.3 times higher than that in healthy subjects.
Renal dysfunction.
In a population pharmacokinetic analysis for patients with ADPKD, tolvaptan concentrations were increased, compared to healthy subjects, as renal function decreased below eGFR of 60 mL/min/1.73 m2. An eGFRCKD-EPI decrease from 72.2 mL/min/1.73 m2 to 9.79 mL/min/1.73 m2 was associated with a 32% reduction in total body clearance.
5.3 Preclinical Safety Data
Genotoxicity.
Tolvaptan tested negative for genotoxicity in in vitro (bacterial reverse mutation assay, mammalian forward mutation assay and chromosomal aberration test in Chinese hamster lung fibroblast cells) and in vivo (rats micronucleus assay) test systems.
Carcinogenicity.
Up to two years of oral administration of tolvaptan to male and female rats at doses up to 1000 mg/kg/day (yielding 1.3 and 3.4-times respectively, the AUC for tolvaptan in patients at the MRHD), to male mice at doses up to 60 mg/kg/day (relative exposure, 0.3) and to female mice at doses up to 100 mg/kg/day (relative exposure, 0.4) did not increase the incidence of tumours. The predictive value of these studies is limited by the inability to obtain high multiples of clinical exposure to tolvaptan. However, negative findings in genotoxicity assays and the absence of pre-neoplastic lesions observed in these and other studies lend support to tolvaptan being unlikely to pose a particular carcinogenic risk in patients.6 Pharmaceutical Particulars
6.1 List of Excipients
Inactive ingredients include maize starch, hyprolose, lactose monohydrate, magnesium stearate, microcrystalline cellulose and ferric oxide as colorant.
6.2 Incompatibilities
See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C. Store in original container.
Protect from light and moisture.
6.5 Nature and Contents of Container
The following pack sizes and configurations are available:
Tolvaptan Lupin 15 mg tablets are available in a PVC/Aluminium blister in packs of 7 or 28 tablets.
Tolvaptan Lupin 30 mg tablets are available in a PVC/Aluminium blister in packs of 7 or 28 tablets.
Tolvaptan Lupin combination pack of 15 mg + 45 mg tablets are available in a PVC/Aluminium foil blister in packs of 56 tablets.
Tolvaptan Lupin combination pack of 30 mg + 60 mg tablets are available in a PVC/Aluminium foil blister in packs of 56 tablets.
Tolvaptan Lupin combination pack of 30 mg + 90 mg tablets are available in a PVC/Aluminium foil blister in packs of 56 tablets.
Not all presentations and pack sizes may be available in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Tolvaptan is practically insoluble in water and the aqueous solubility of the drug substance is poor (~ 0.1 mg/250 mL) across all pH ranges. It is slightly soluble in ethyl acetate, sparingly soluble in ethanol, soluble in methanol and freely soluble in benzyl alcohol. The octanol: water partition coefficient was reported to be greater than 5000 at 25°C.
Chemical structure.
 Chemical name: (±)-4'-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl) carbonyl]-o-tolu-m-toluidide.
Chemical name: (±)-4'-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl) carbonyl]-o-tolu-m-toluidide.
Molecular formula: C26H25ClN2O3.
Molecular weight: 448.94.
CAS number.
150683-30-0.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes
