What is in this leaflet
This leaflet answers some common questions about TREMFYA (pronounced trem-fye-ah). It does not contain all the available information. It does not take the place of talking to your doctor, nurse or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using TREMFYA against the benefits it is expected to have for you.
If you have any concerns about using this medicine, ask your doctor, nurse or pharmacist.
Keep this leaflet. You may need to read it again.
What TREMFYA is used for
TREMFYA contains the active substance guselkumab which is a type of protein called a monoclonal antibody. This medicine works by neutralising the activity of a protein called IL-23, which is present in increased levels in people with psoriasis.
TREMFYA is used to treat:
- adults with moderate to severe plaque psoriasis, an inflammatory condition affecting the skin and nails. TREMFYA can improve skin clearance and nail appearance and reduce symptoms of psoriasis, such as scaling, shedding, flaking, itching, pain, and burning.
- adults with active psoriatic arthritis, an inflammatory disease of the joints in which psoriasis usually occurs in association with arthritis. If you have active psoriatic arthritis, you will be given TREMFYA alone or in combination with a conventional Disease Modifying Anti-Rheumatic Drug (DMARD) such as methotrexate.
Ask your doctor if you have any questions about why TREMFYA has been prescribed for you. Your doctor may have prescribed TREMFYA for another reason.
Before you use TREMFYA
When you must not use it
Do not use TREMFYA if:
- You have an allergy to guselkumab (the active ingredient in the medicine) or to any of the ingredients listed at the end of this leaflet.
Symptoms of an allergic reaction may include rash, itching or hives on the skin, shortness of breath, wheezing or difficulty breathing, a tight feeling in your chest, swelling of the face, lips, tongue or other parts of the body.
- The packaging is torn or shows signs of tampering.
- The expiry date on the pack has passed.
Before you start to use it
Tell your doctor if you:
- are being treated for an infection
- have an infection that does not go away or keeps coming back
- have tuberculosis (TB) or have been in close contact with someone with TB
- think you have an infection or have symptoms of an infection such as:
- fever or flu-like symptoms
- blood in your phlegm (mucus)
- muscle aches
- cough
- shortness of breath
- weight loss
- diarrhoea or stomach pain
- burning when you urinate or urinating more often than normal
- warm, red, or painful skin or sores on your body different from your psoriasis
After starting TREMFYA, talk to your doctor straight away if you have any of the symptoms of infection listed above.
TREMFYA may lower your ability to fight infections and may increase your risk of infections. Do not use TREMFYA if you have any symptoms of infection unless you are instructed to by your doctor. - have recently had a vaccination or if you are due to have a vaccination during treatment with TREMFYA.
You should not be given certain types of vaccines (live vaccines) while using TREMFYA.
If you are not sure if any of the above applies to you, talk to your doctor, pharmacist or nurse before using TREMFYA. - allergic reactions
Tell your doctor or get emergency medical help right away if you think you are having an allergic reaction. - are pregnant or plan to become pregnant.
The effects of this medicine in pregnant women are not known. Talk to your doctor if you are pregnant, think you may be pregnant or are planning to have a baby.
If you are a woman of childbearing potential, use adequate contraception while using TREMFYA and for at least 12 weeks after the last TREMFYA dose. Talk to your doctor about your contraception options. - are breastfeeding or planning to breastfeed. You and your doctor should decide if you will breastfeed while using TREMFYA.
If you have not told your doctor about any of the above, tell them before you start using TREMFYA.
Your doctor will discuss with you the benefits of using it against the potential risks.
Taking or being given other medicines
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements.
How to use TREMFYA
TREMFYA is only available on prescription.
Always use this medicine exactly as your doctor has told you. Check with your doctor, nurse or pharmacist if you are not sure.
TREMFYA is given by injection under your skin (subcutaneous injection). At the start of your therapy, TREMFYA may be injected by your healthcare provider. Your doctor or nurse may decide that it is right for you to learn how to inject TREMFYA yourself.
It is important not to try to inject yourself until you have been trained by a healthcare professional. If you have not been trained, please contact your healthcare provider to schedule a training session. A caregiver may also give you your TREMFYA injection after proper training.
Before use, remove the carton from the refrigerator and keep the pre-filled syringe inside the carton and allow to reach room temperature by waiting for 30 minutes.
Read the “Instructions for Use” leaflet for the pre-filled syringe or pre-filled pen carefully before using TREMFYA.
Nurse support is available by calling the Janssen Immunology Patient Support Program on 1800 666 845.
How much to use
Your doctor will decide how much TREMFYA you need and for how long.
For plaque psoriasis
- The first dose is 100 mg (the content of 1 pre-filled syringe or 1 pre-filled pen) by subcutaneous injection. This may be given by your doctor or nurse.
- After the first dose, you will have the next dose 4 weeks later, and then every 8 weeks. Subsequent doses are the same as the first dose (the content of 1 pre-filled syringe or 1 pre-filled pen).
For psoriatic arthritis
- The first dose is 100 mg (the content of 1 pre-filled syringe or 1 pre filled pen) by subcutaneous injection. This may be given by your doctor or nurse.
- After the first dose, you will have the next dose 4 weeks later, and then every 8 weeks. Subsequent doses are the same as the first dose (the content of 1 pre-filled syringe or 1 pre-filled pen).
- TREMFYA can be used with or without a type of medicine called a conventional Disease-Modifying Anti-Rheumatic Drug (DMARD), such as methotrexate.
You should not stop using TREMFYA without speaking to your doctor first. If you stop treatment, symptoms of psoriasis may come back.
If you forget to use it
If you forget to take your TREMFYA dose, inject a dose as soon as you remember.
Then, take your next dose at your regular scheduled time
Do not use a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your pharmacist, nurse or doctor.
If you use too much (overdose)
If you have received more TREMFYA than you should, or the dose has been given sooner than prescribed, immediately telephone your doctor or the Poisons Information Centre for advice, or go to Accident and Emergency at your nearest hospital.
Do this even if there are no signs of discomfort or poisoning.
You may need urgent medical attention.
Poisons Information Centre telephone number:
- Australia: 13 11 26
While you are using TREMFYA
Things you must do
Tell any other doctors, dentists and pharmacists who are treating you that you are using TREMFYA.
If you are about to start taking a new medicine, tell your doctor and pharmacist that you are taking TREMFYA.
Tell your doctor, nurse or pharmacist if the medicine starts to upset you or your symptoms become worse.
Tell your doctor immediately if you develop any symptoms of an infection (see “Before You Use TREMFYA”).
Tell your doctor if you become pregnant while using TREMFYA.
Things you must not do
You should not receive a live vaccine while taking TREMFYA. Talk to your doctor, pharmacist or nurse before receiving any vaccination while taking TREMFYA.
Do not give TREMFYA to anyone else, even if they have the same condition as you.
Do not stop using TREMFYA, or change the dose, unless your doctor tells you to.
Things to be careful of
Be careful driving or operating machinery until you know how TREMFYA affects you. TREMFYA should not affect your ability to drive or use machines. However, make sure you know how you react to it before you do anything that could be dangerous if you feel dizzy.
Side effects
All medicines can have some side effects. Sometimes they are serious, but most of the time they are not. Your doctor has weighed the risks of using this medicine against the benefits they expect it will have for you.
Tell your doctor immediately if you experience any of the following side effects:
- cold and/or flu symptoms, chest infection (upper respiratory infection)
- Headache
- joint pain (arthralgia)
- stomach flu (gastroenteritis)
- diarrhoea
- redness at the injection site
- pain at the injection site
- cold sores (herpes simplex infections)
- tinea
- blood tests may show decreased number of a type of white blood cell called neutrophils or increased level of liver enzymes.
Tell your doctor if you experience any side effects even if they are not on this list or do not feel well.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Stop taking TREMFYA and tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you have any signs of an allergic reaction. You need urgent medical attention. Signs of an allergic reaction may include rash, itching or hives on the skin, shortness of breath, wheezing or difficulty breathing, a tight feeling in your chest, swelling of the face, lips, tongue or other parts of the body.
Do not be alarmed by the list of possible side effects. You may not experience any of them.
Ask your doctor, nurse or pharmacist any questions you may have.
How to store TREMFYA
Storage
Store TREMFYA in a refrigerator between 2°C and 8°C.
TREMFYA should not be frozen.
Keep the product in the original carton to protect from light until the time of use.
Do not shake TREMFYA.
Before use, remove the carton from the refrigerator and keep the pre-filled syringe or pre-filled pen inside the carton and allow to reach room temperature by waiting for 30 minutes.
Always keep medicine out of the reach of children.
After using TREMFYA
Disposal
After injecting TREMFYA, place the used syringe or pen immediately into a sharps container. Do not put the used syringe or pen into your normal household or recycling waste.
The syringe or pen should never be re-used. Discard any unused portions of TREMFYA.
If your doctor tells you to stop taking TREMFYA or it has passed its expiry date, ask your pharmacist what to do with any that is left over.
Product description
What it looks like
TREMFYA is a clear, colourless to light yellow solution and may contain clear or white particles of protein. This appearance is not unusual for solutions containing proteins.
Do not use TREMFYA if the solution is discoloured, cloudy, or you can see other particulate matter floating in it.
TREMFYA is available as a single use pre-filled syringe or pre-filled pen (One-Press® patient controlled injector) in a carton. Each carton contains 1 pre-filled syringe or 1 prefilled pen.
An instruction for use leaflet explaining how to self-administer the product is included in the pack.
Do not use TREMFYA if the packaging is torn or shows signs of tampering.
Ingredients
The active ingredient is guselkumab.
The inactive ingredients are histidine, histidine hydrochloride monohydrate, polysorbate 80, sucrose, and water for injection.
No preservatives are present.
Sponsor
JANSSEN-CILAG Pty Ltd
1-5 Khartoum Road
Macquarie Park NSW 2113 Australia
Telephone: 1800 226 334
Australian Registration Number
100 mg pre-filled syringe: AUST R 286020
100 mg pre-filled pen: AUST R 321410
Date of Preparation
May 2022
Nurse and educational support
The Janssen Immunology Patient Support Program is available to patients prescribed TREMFYA. It offers:
- starter kit
- one-to-one nurse support
- reminder service
- ongoing education
- wellbeing support
Call 1800 666 845
Published by MIMS July 2022






 In pooled phase 2/3 Crohn's disease clinical studies, through the reporting period of approximately one-year, adverse events of increased transaminases (includes ALT increased, AST increased, hepatic enzyme increased, transaminases increased, hepatic function abnormal, and liver function test increased) were reported in 3.4% of patients in the Tremfya 200 mg SC q4w treatment group and 4.1% of patients in the Tremfya 100 mg SC q8w treatment group compared to 2.4% in the placebo group.
In pooled phase 2/3 Crohn's disease clinical studies, through the reporting period of approximately one-year, adverse events of increased transaminases (includes ALT increased, AST increased, hepatic enzyme increased, transaminases increased, hepatic function abnormal, and liver function test increased) were reported in 3.4% of patients in the Tremfya 200 mg SC q4w treatment group and 4.1% of patients in the Tremfya 100 mg SC q8w treatment group compared to 2.4% in the placebo group.
 Response over time.Guselkumab demonstrated rapid onset of efficacy, with a significantly higher percent improvement in PASI as compared with placebo as early as Week 2 (p < 0.001). The percentage of subjects achieving a PASI 90 response was numerically higher for guselkumab than adalimumab starting at Week 8 with the difference reaching a maximum around Week 20 (VOYAGE 1 and 2) and maintained through Week 48 (VOYAGE 1). In VOYAGE 1, for subjects receiving continuous guselkumab treatment, PASI 90 response was maintained from Week 52 to Week 252 with 84.1% (329/391) of patients continuing to achieve a PASI 90 response at Week 252 (see Figures 1 and 2).
Response over time.Guselkumab demonstrated rapid onset of efficacy, with a significantly higher percent improvement in PASI as compared with placebo as early as Week 2 (p < 0.001). The percentage of subjects achieving a PASI 90 response was numerically higher for guselkumab than adalimumab starting at Week 8 with the difference reaching a maximum around Week 20 (VOYAGE 1 and 2) and maintained through Week 48 (VOYAGE 1). In VOYAGE 1, for subjects receiving continuous guselkumab treatment, PASI 90 response was maintained from Week 52 to Week 252 with 84.1% (329/391) of patients continuing to achieve a PASI 90 response at Week 252 (see Figures 1 and 2).
 The efficacy and safety of guselkumab was demonstrated regardless of age, gender, race, body weight, plaques location, PASI baseline severity, concurrent psoriatic arthritis, and previous treatment with a biologic therapy. Guselkumab was efficacious in conventional systemic-naive, biologic-naive, and biologic-exposed patients.
The efficacy and safety of guselkumab was demonstrated regardless of age, gender, race, body weight, plaques location, PASI baseline severity, concurrent psoriatic arthritis, and previous treatment with a biologic therapy. Guselkumab was efficacious in conventional systemic-naive, biologic-naive, and biologic-exposed patients.

 Tremfya and secukinumab PASI 90 response rates through Week 48 are presented in Figure 4.
Tremfya and secukinumab PASI 90 response rates through Week 48 are presented in Figure 4.
 The baseline disease characteristics were generally comparable across treatment groups with median baseline BSA of 20%, median baseline PASI score approximately 17, and baseline IGA score of severe for 20% and 24% of subjects, and a history of psoriatic arthritis for < 5% of subjects.
The baseline disease characteristics were generally comparable across treatment groups with median baseline BSA of 20%, median baseline PASI score approximately 17, and baseline IGA score of severe for 20% and 24% of subjects, and a history of psoriatic arthritis for < 5% of subjects.

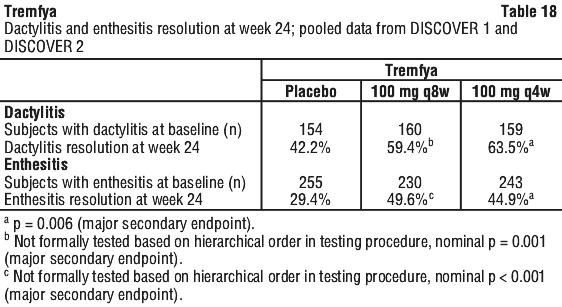
 Clinical response was maintained up to Week 52 as assessed by ACR 20/50/70, DAS 28 (CRP), MDA, IGA and PASI 90 response rates (see Table 16).
Clinical response was maintained up to Week 52 as assessed by ACR 20/50/70, DAS 28 (CRP), MDA, IGA and PASI 90 response rates (see Table 16).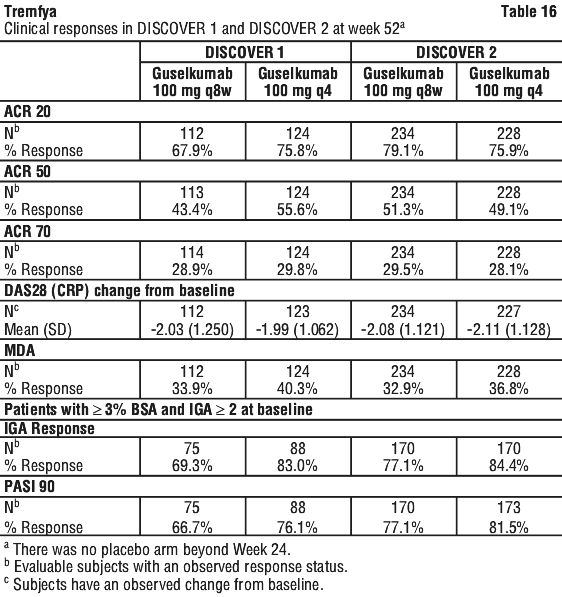 In DISCOVER 1 and 2, patients treated with Tremfya who had spondylitis with peripheral arthritis as their primary presentation, demonstrated greater improvement from baseline in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) compared to placebo at Week 24. Improvement in BASDAI was maintained from Week 24 to Week 52 in DISCOVER 1 and Week 100 in DISCOVER 2.
In DISCOVER 1 and 2, patients treated with Tremfya who had spondylitis with peripheral arthritis as their primary presentation, demonstrated greater improvement from baseline in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) compared to placebo at Week 24. Improvement in BASDAI was maintained from Week 24 to Week 52 in DISCOVER 1 and Week 100 in DISCOVER 2. In DISCOVER 2, for subjects receiving continuous guselkumab treatment at week 24, ACR 20 response was maintained from Week 24 to Week 52 (see Figure 6). For subjects receiving continuous guselkumab treatment at week 52, ACR 20 response was maintained from Week 52 to Week 100 (see Figure 7).
In DISCOVER 2, for subjects receiving continuous guselkumab treatment at week 24, ACR 20 response was maintained from Week 24 to Week 52 (see Figure 6). For subjects receiving continuous guselkumab treatment at week 52, ACR 20 response was maintained from Week 52 to Week 100 (see Figure 7).
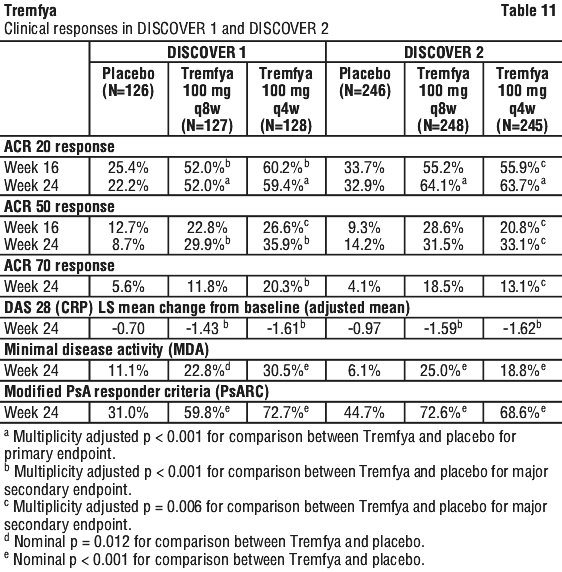
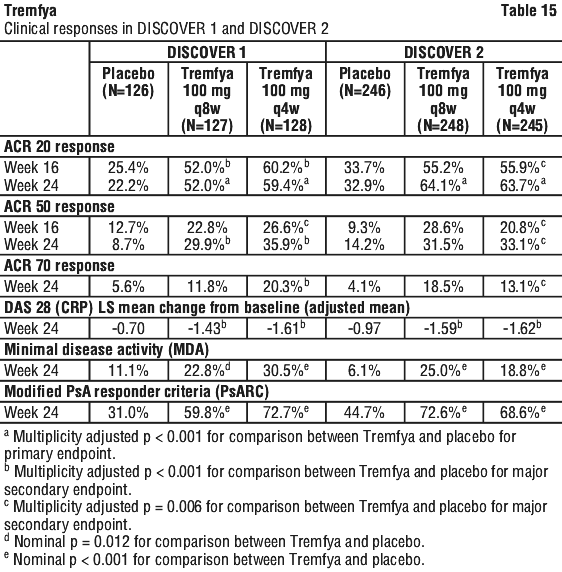
 In DISCOVER 1 and DISCOVER 2, median percent improvement from baseline was observed in all components of the ACR response criteria.
In DISCOVER 1 and DISCOVER 2, median percent improvement from baseline was observed in all components of the ACR response criteria.
 The mean change from baseline in total modified vdH-S was similar in the guselkumab q8w and q4w groups at Week 52 (0.97 and 1.07, respectively) and at Week 100 (1.50 and 1.68 respectively).
The mean change from baseline in total modified vdH-S was similar in the guselkumab q8w and q4w groups at Week 52 (0.97 and 1.07, respectively) and at Week 100 (1.50 and 1.68 respectively).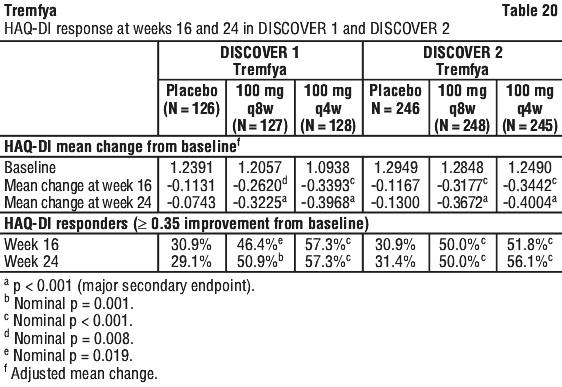 At Week 24, subjects in both the Tremfya 100 mg q8w and q4w dose groups in both DISCOVER 1 and DISCOVER 2 showed greater improvement from baseline in the SF-36 PCS with no worsening in the SF-36 MCS compared with placebo. At Week 24 there was consistent evidence of effect in the physical functioning, role-physical, bodily-pain, general health, social-functioning and vitality domains but not in the role-emotional and mental health domains. At Week 24, a greater mean increase from baseline in Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) score was observed in guselkumab treated patients compared to placebo in both DISCOVER 1 (5.609, 5.841 and 2.206 in the 100 mg q8w, 100 mg q4w and placebo groups, respectively; both nominal p < 0.001) and DISCOVER 2 (7.550, 7.111 and 3.559 in the 100 mg q8w, 100 mg q4w and placebo groups, respectively; both nominal p < 0.001). In DISCOVER 2, greater improvements in health-related quality of life as measured by mean change from baseline in the Dermatology Life Quality Index (DLQI) were observed in guselkumab treated patients compared to placebo at Week 24 (-8.954, -8.853, and -2.129 in the 100 mg q8w, 100 mg q4w and placebo groups, respectively; both nominal p < 0.001). In DISCOVER 2, greater improvements were also observed in overall work impairment and activity impairment as assessed by the Work Productivity and Activity Impairment (WPAI)-PsA questionnaire compared to placebo at Week 24. Improvements in SF-36 PCS, SF-36 MCS, FACIT-F, DLQI and WPAI-PsA scores were maintained from Week 24 to Week 52 in DISCOVER 1 and Week 100 in DISCOVER 2.
At Week 24, subjects in both the Tremfya 100 mg q8w and q4w dose groups in both DISCOVER 1 and DISCOVER 2 showed greater improvement from baseline in the SF-36 PCS with no worsening in the SF-36 MCS compared with placebo. At Week 24 there was consistent evidence of effect in the physical functioning, role-physical, bodily-pain, general health, social-functioning and vitality domains but not in the role-emotional and mental health domains. At Week 24, a greater mean increase from baseline in Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) score was observed in guselkumab treated patients compared to placebo in both DISCOVER 1 (5.609, 5.841 and 2.206 in the 100 mg q8w, 100 mg q4w and placebo groups, respectively; both nominal p < 0.001) and DISCOVER 2 (7.550, 7.111 and 3.559 in the 100 mg q8w, 100 mg q4w and placebo groups, respectively; both nominal p < 0.001). In DISCOVER 2, greater improvements in health-related quality of life as measured by mean change from baseline in the Dermatology Life Quality Index (DLQI) were observed in guselkumab treated patients compared to placebo at Week 24 (-8.954, -8.853, and -2.129 in the 100 mg q8w, 100 mg q4w and placebo groups, respectively; both nominal p < 0.001). In DISCOVER 2, greater improvements were also observed in overall work impairment and activity impairment as assessed by the Work Productivity and Activity Impairment (WPAI)-PsA questionnaire compared to placebo at Week 24. Improvements in SF-36 PCS, SF-36 MCS, FACIT-F, DLQI and WPAI-PsA scores were maintained from Week 24 to Week 52 in DISCOVER 1 and Week 100 in DISCOVER 2.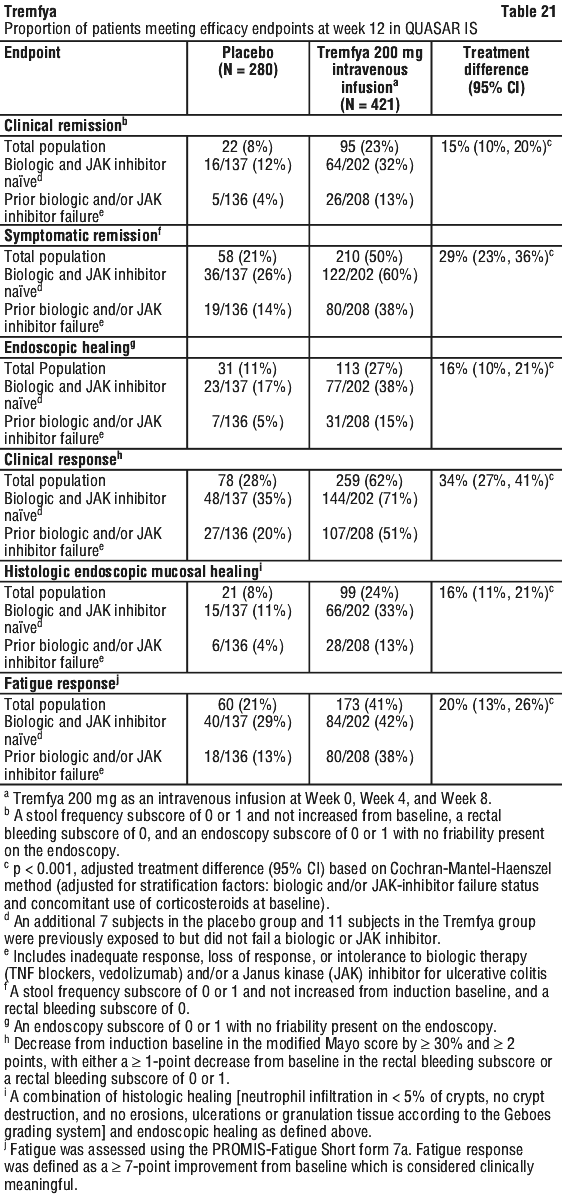 QUASAR IS and QUASAR induction dose-ranging study also enrolled patients with a baseline mMS of 4, including an ES of 2 or 3 and a RBS ≥ 1. In these patients, Tremfya efficacy relative to placebo, as measured by clinical remission, clinical response, and endoscopic healing at Week 12, was consistent with the total moderately to severely active UC population.
QUASAR IS and QUASAR induction dose-ranging study also enrolled patients with a baseline mMS of 4, including an ES of 2 or 3 and a RBS ≥ 1. In these patients, Tremfya efficacy relative to placebo, as measured by clinical remission, clinical response, and endoscopic healing at Week 12, was consistent with the total moderately to severely active UC population.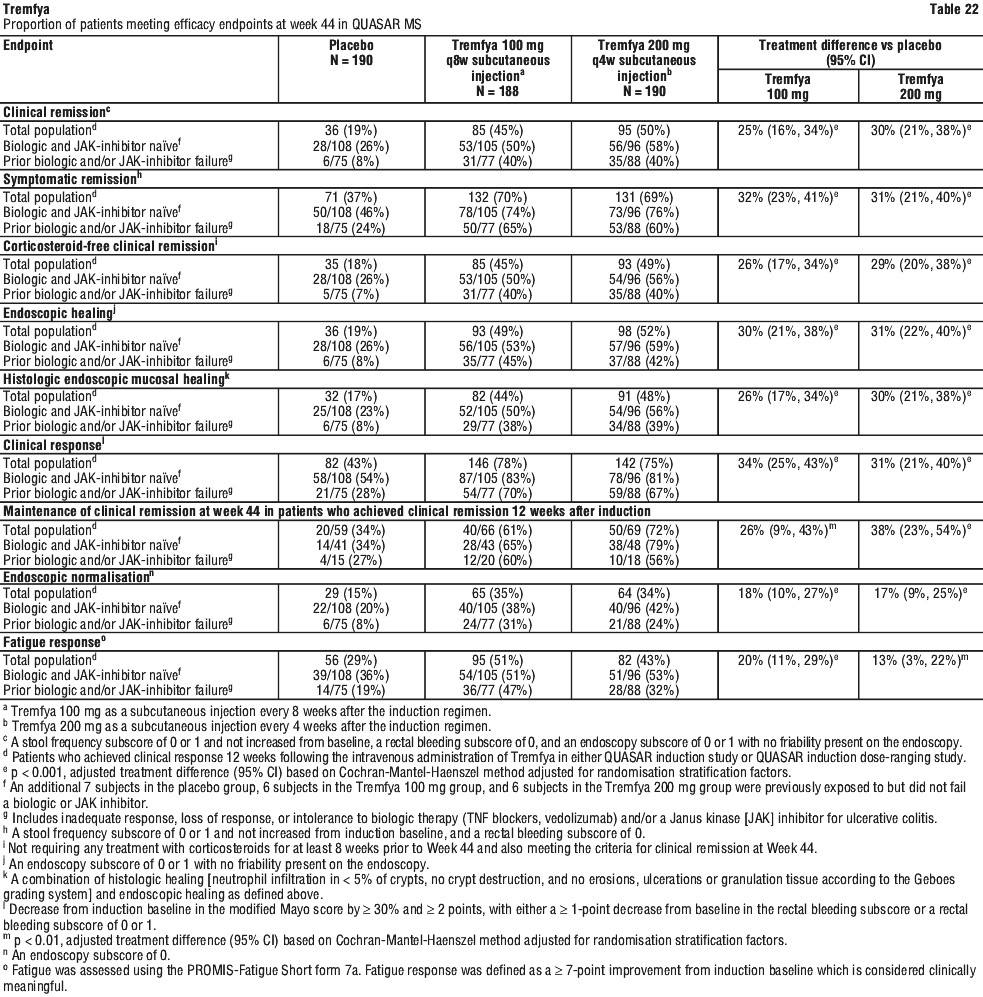 In QUASAR IS and QUASAR MS, the efficacy and safety of Tremfya was consistently demonstrated regardless of age, sex, race, body weight, and previous treatment with a biologic therapy or JAK inhibitor. Tremfya was efficacious in biologic and JAK inhibitor naïve patients, as well as in patients who previously failed a biologic and/or JAK inhibitor.
In QUASAR IS and QUASAR MS, the efficacy and safety of Tremfya was consistently demonstrated regardless of age, sex, race, body weight, and previous treatment with a biologic therapy or JAK inhibitor. Tremfya was efficacious in biologic and JAK inhibitor naïve patients, as well as in patients who previously failed a biologic and/or JAK inhibitor.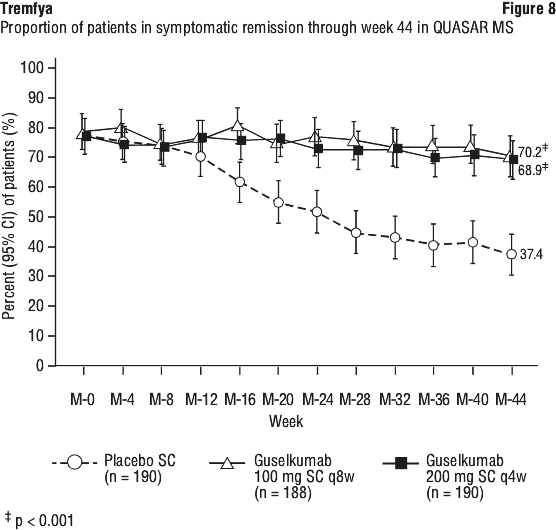
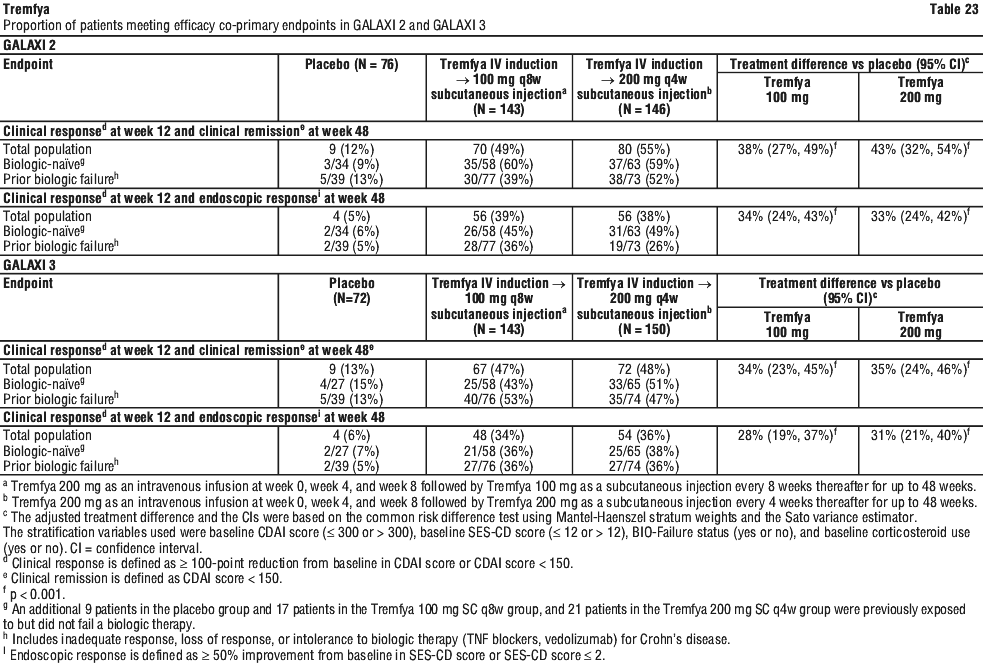
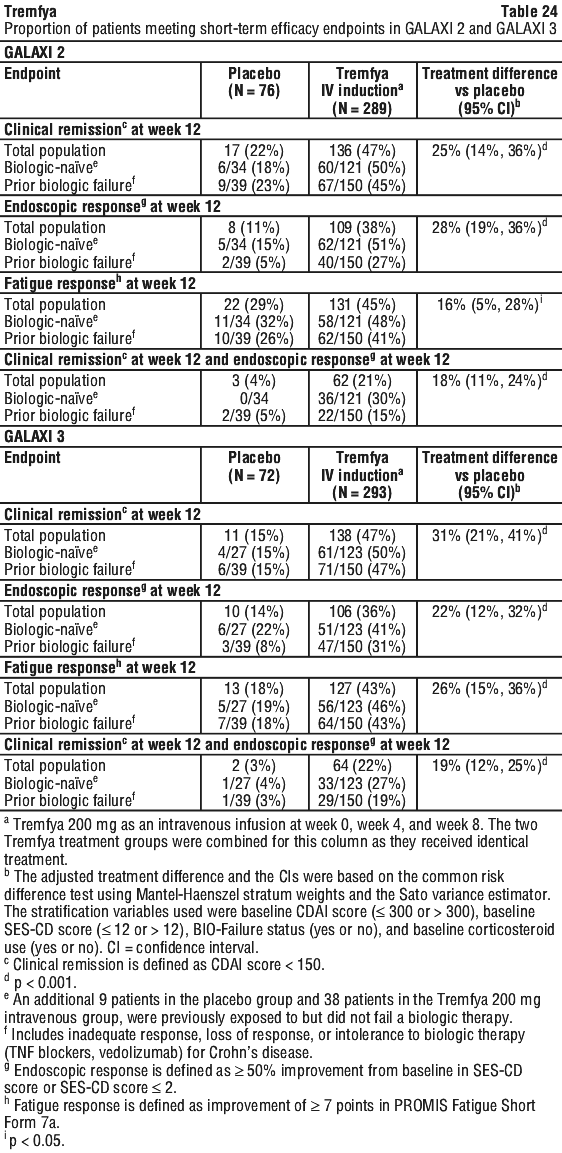 In GALAXI 2 and GALAXI 3, a greater proportion of patients achieved clinical response (assessed by CDAI) at Week 4 as well as endoscopic remission at Week 12 in patients treated with Tremfya 200 mg intravenous induction compared to placebo.
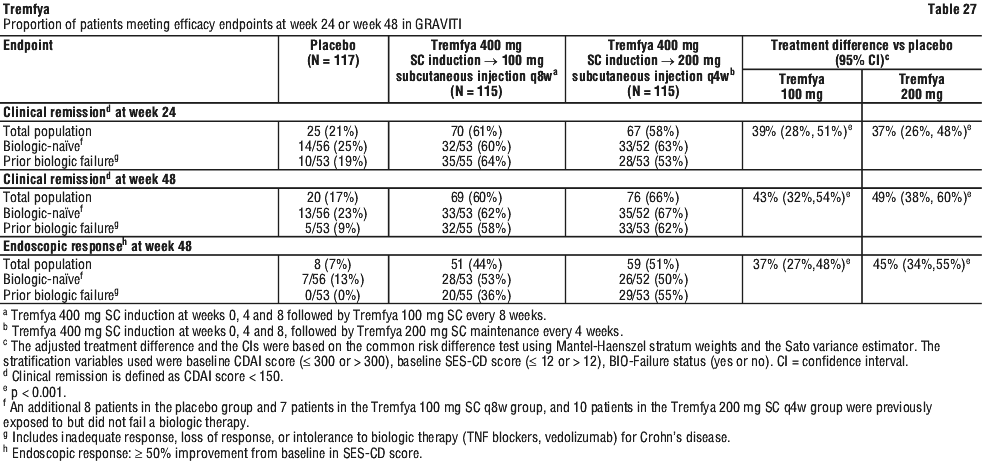
In GALAXI 2 and GALAXI 3, a greater proportion of patients achieved clinical response (assessed by CDAI) at Week 4 as well as endoscopic remission at Week 12 in patients treated with Tremfya 200 mg intravenous induction compared to placebo.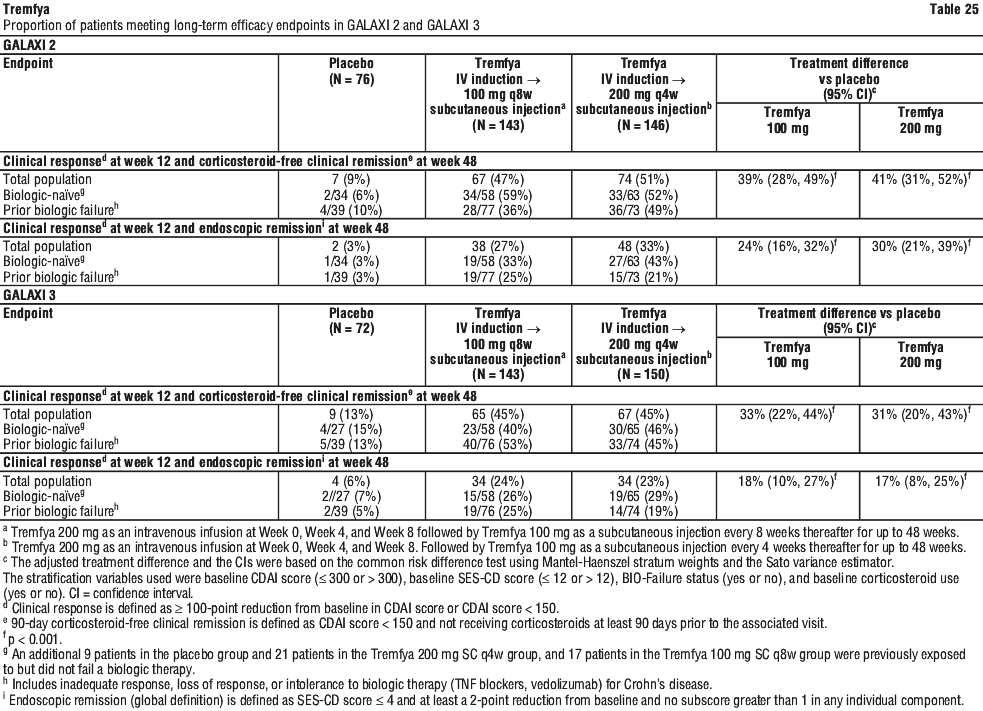 In the pooled GALAXI Phase 3 studies subpopulation analysis, patients with high inflammatory burden after completion of induction dosing derived additional benefit from Tremfya 200 mg subcutaneous q4w compared to the 100 mg subcutaneous q8w. A clinically meaningful difference of 14 to 17 percentage points was observed between the two Tremfya dose groups among patients with a CRP level of > 5 mg/L after completion of induction, for the endpoints of clinical remission at Week 48 (100 mg subcutaneous q8w: 54% vs 200 mg subcutaneous q4w: 71%); and endoscopic response at Week 48 (100 mg subcutaneous q8w: 36% vs 200 mg subcutaneous q4w: 50%).
In the pooled GALAXI Phase 3 studies subpopulation analysis, patients with high inflammatory burden after completion of induction dosing derived additional benefit from Tremfya 200 mg subcutaneous q4w compared to the 100 mg subcutaneous q8w. A clinically meaningful difference of 14 to 17 percentage points was observed between the two Tremfya dose groups among patients with a CRP level of > 5 mg/L after completion of induction, for the endpoints of clinical remission at Week 48 (100 mg subcutaneous q8w: 54% vs 200 mg subcutaneous q4w: 71%); and endoscopic response at Week 48 (100 mg subcutaneous q8w: 36% vs 200 mg subcutaneous q4w: 50%).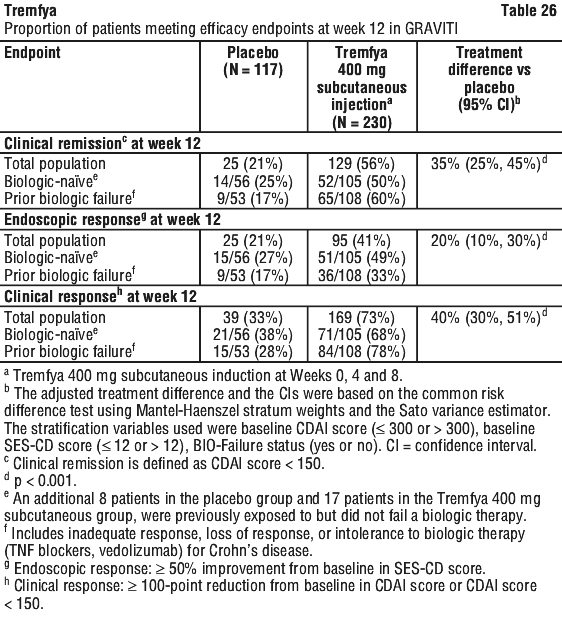 Onset of clinical response and clinical remission based on CDAI occurred as early as Week 4 in a greater proportion of patients treated with the Tremfya induction regimen compared to placebo.
Onset of clinical response and clinical remission based on CDAI occurred as early as Week 4 in a greater proportion of patients treated with the Tremfya induction regimen compared to placebo.