1 Name of Medicine
Elexacaftor, tezacaftor and ivacaftor in combination, and ivacaftor.
2 Qualitative and Quantitative Composition
Tablets.
Trikafta 100/50/75 film-coated tablets.
One morning dose film-coated tablet contains elexacaftor 100 mg, tezacaftor 50 mg and ivacaftor 75 mg.
One evening dose film-coated tablet contains ivacaftor 150 mg.
Excipients with known effect: contains sugars as lactose (150 mg ivacaftor tablet).
For the full list of excipients, see Section 6.1 List of Excipients.
Trikafta 50/25/37.5 film-coated tablets.
One morning dose film-coated tablet contains elexacaftor 50 mg, tezacaftor 25 mg and ivacaftor 37.5 mg.
One evening dose film-coated tablet contains ivacaftor 75 mg.
Excipients with known effect: contains sugars as lactose (75 mg ivacaftor tablet).
For the full list of excipients, see Section 6.1 List of Excipients.
Granules.
Trikafta 100/50/75 granules.
One morning dose sachet contains elexacaftor 100 mg, tezacaftor 50 mg and ivacaftor 75 mg.
One evening dose sachet contains ivacaftor 75 mg.
Excipients with known effect: contains sugars as lactose, contains sucralose.
For the full list of excipients, see Section 6.1 List of Excipients.
Trikafta 80/40/60 granules.
One morning dose sachet contains elexacaftor 80 mg, tezacaftor 40 mg and ivacaftor 60 mg.
One evening dose sachet contains ivacaftor 59.5 mg.
Excipients with known effect: contains sugars as lactose, contains sucralose.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Composite pack.
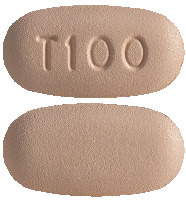
Trikafta 100/50/75 film-coated tablets. Elexacaftor/tezacaftor/ivacaftor 100 mg/50 mg/75 mg tablet.
Orange, capsule shaped tablet with "T100" debossed on one side and plain on the other (7.9 mm x 15.5 mm).
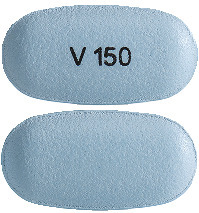
Ivacaftor 150 mg tablet.
Light blue, capsule-shaped tablet printed with "V 150" in black ink on one side and plain on the other (16.5 mm x 8.4 mm).
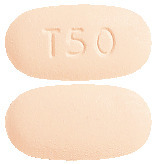
Trikafta 50/25/37.5 film-coated tablets. Elexacaftor/tezacaftor/ivacaftor 50 mg/25 mg/37.5 mg tablet.
Light orange, capsule shaped tablet with "T50" debossed on one side and plain on the other (6.4 mm x 12.2 mm).
Ivacaftor 75 mg tablet.
Light blue, capsule shaped tablet printed with "V 75" in black ink on one side and plain on the other (12.7 mm x 6.8 mm).
Trikafta 100/50/75 granules and 80/40/60 granules. Elexacaftor/tezacaftor/ivacaftor 100 mg/50 mg/75 mg and 80 mg/40 mg/60 mg granules.
White to off-white, sweetened, unflavored granules approximately 2 mm in diameter.
Ivacaftor 75 mg granules and 59.5 mg granules.
White to off-white, sweetened, unflavored granules approximately 2 mm in diameter.4.1 Therapeutic Indications
Trikafta is indicated for the treatment of those who meet the diagnostic criteria of cystic fibrosis (CF) in patients aged 2 years and older who have at least one mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that is responsive based on clinical or in vitro evidence (see Section 5.1 Pharmacodynamic Properties).
4.2 Dose and Method of Administration
Trikafta should only be prescribed by physicians with experience in the treatment of CF.
Dosage.
Adults and paediatric patients aged 2 years and older should be dosed according to Table 1.
 The morning and evening dose should be taken with fat containing food, approximately 12 hours apart.
The morning and evening dose should be taken with fat containing food, approximately 12 hours apart.
Missed dose.
If 6 hours or less have passed since the missed morning or evening dose, the patient should take the missed dose as soon as possible and continue on the original schedule.
If more than 6 hours have passed since:
the missed morning dose, the patient should take the missed dose as soon as possible and should not take the evening dose. The next scheduled morning dose should be taken at the usual time;
the missed evening dose, the patient should not take the missed dose. The next scheduled morning dose should be taken at the usual time.
Morning and evening doses should not be taken at the same time.
Method of administration.
A fat-containing meal or snack should be consumed just before or just after dosing of Trikafta. Meals and snacks recommended in CF guidelines or meals recommended in standard nutritional guidelines contain adequate amounts of fat. A serving size of foods appropriate for age from a recommended CF diet should be given. Examples of meals or snacks that contain fat are those prepared with butter or oils or those containing eggs, cheeses, nuts, chocolate, whole milk, whole-milk dairy products, meats, avocado, hummus, oily fish, and soy-based products (tofu) (see Section 5.2 Pharmacokinetic Properties).
Food or drink containing grapefruit should be avoided during treatment with Trikafta (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Tablets.
For oral use. Patients should be instructed to swallow the tablets whole.
Granules.
For oral use. The entire contents of each sachet of granules should be mixed with one teaspoon (5 mL) of age-appropriate soft food or liquid and the mixture completely consumed. Food or liquid should be at room temperature or below. Each sachet is for single use only. Once mixed, the product has been shown to be stable for one hour, and therefore should be ingested during this period. Some examples of soft food or liquids include pureed fruits or vegetables, yogurt, applesauce, water, milk, or juice. A fat-containing meal or snack should be consumed just before or after dosing.
Dosage adjustment.
Hepatic impairment.
Treatment of patients with moderate hepatic impairment (Child-Pugh Class B) is not recommended. Treatment of patients with moderate hepatic impairment should only be considered when there is a clear medical need and the benefits are expected to outweigh the risks. If used, Trikafta should be used with caution at a reduced dose (see Table 2).
Studies have not been conducted in patients with severe hepatic impairment (Child-Pugh Class C), but the exposure is expected to be higher than in patients with moderate hepatic impairment. Patients with severe hepatic impairment should not be treated with Trikafta.
No dose adjustment is recommended for patients with mild hepatic impairment (Child-Pugh Class A) (see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects); Section 5.2 Pharmacokinetic Properties).

Renal impairment.
No dose adjustment is recommended for patients with mild and moderate renal impairment. Caution is recommended for patients with severe renal impairment or end-stage renal disease (see Section 5.2 Pharmacokinetic Properties).
Concomitant use of CYP3A inhibitors.
When co-administered with moderate CYP3A inhibitors (e.g. fluconazole, erythromycin, verapamil) or strong CYP3A inhibitors (e.g. ketoconazole, itraconazole, posaconazole, voriconazole, telithromycin, and clarithromycin), the dose should be reduced as in Table 3 (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).

4.3 Contraindications
In cases of hypersensitivity to the active substance or to any component of this medication, patients should not be treated with this medicine.
4.4 Special Warnings and Precautions for Use
Use in hepatic impairment.
Patients with severe hepatic impairment (Child-Pugh Class C) should not be treated with Trikafta. Treatment of patients with moderate hepatic impairment (Child-Pugh Class B) is not recommended. For patients with moderate hepatic impairment, Trikafta should only be used if there is a clear medical need and the benefits are expected to outweigh the risks. No dose adjustment is recommended for patients with mild hepatic impairment (Child-Pugh Class A) (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Elevated transaminases and hepatic injury.
Cases of liver failure leading to transplantation have been reported within the first 6 months of treatment in patients with and without pre-existing advanced liver disease.
Elevated transaminases are common in patients with CF and have been observed in patients treated with Trikafta. In some instances, these elevations have been associated with concomitant elevations in total bilirubin. Assessments of transaminases (ALT and AST) and total bilirubin are recommended for all patients prior to initiating Trikafta, every month during the first 6 months of treatment, every 3 months during the next 6 months and annually thereafter. For patients with a history of liver disease or transaminase elevations, more frequent monitoring should be considered.
Interrupt Trikafta and promptly measure serum transaminases and total bilirubin if a patient develops clinical signs or symptoms suggestive of liver injury (e.g. jaundice and/or dark urine, unexplained nausea or vomiting, right upper quadrant pain, or anorexia). Interrupt dosing in the event of ALT or AST > 5 x the upper limit of normal (ULN), or ALT or AST > 3 x ULN with total bilirubin > 2 x ULN. Follow laboratory tests closely until the abnormalities resolve.
Following resolution, consider the benefits and risks of resuming treatment (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects); Section 5.2 Pharmacokinetic Properties). Patients who resume treatment after interruption should be monitored closely.
In patients with pre-existing advanced liver disease (e.g. cirrhosis, portal hypertension), Trikafta should be used with caution and only if the benefits are expected to outweigh the risks (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects); Section 5.2 Pharmacokinetic Properties).
Interactions with medicinal products.
CYP3A inducers.
Exposure to ivacaftor is significantly decreased and exposures to elexacaftor and tezacaftor are expected to decrease by the concomitant use of CYP3A inducers, potentially resulting in the reduction of Trikafta efficacy; therefore, co-administration with strong CYP3A inducers is not recommended (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
CYP3A inhibitors.
Exposure to elexacaftor, tezacaftor and ivacaftor are increased when co-administered with moderate or strong CYP3A inhibitors. Therefore, the dose of Trikafta should be reduced when used concomitantly with moderate or strong CYP3A inhibitors (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 4.2 Dose and Method of Administration, Table 3).
Hypersensitivity reactions, including anaphylaxis.
Hypersensitivity reactions, including cases of angioedema and anaphylaxis, have been reported in the post-marketing setting. If signs or symptoms of serious hypersensitivity reactions develop during treatment, discontinue Trikafta and institute appropriate therapy. Consider the benefits and risks for the individual patient to determine whether to resume treatment with Trikafta.
Cataracts.
Cases of non-congenital lens opacities without impact on vision have been reported in paediatric patients treated with ivacaftor containing regimens. Although other risk factors were present in some cases (such as corticosteroid use, exposure to radiation) a possible risk attributable to treatment with ivacaftor cannot be excluded. Baseline and follow-up ophthalmological examinations are recommended in paediatric patients initiating treatment with Trikafta. Cataracts were seen in juvenile rats treated with ivacaftor from postnatal Day 7 through 35 at oral dose levels of 10 mg/kg/day and higher (yielding systemic exposure in animals approximately 5 times lower than that in patients at the maximum recommended human dose [MRHD] based on summed AUCs of the ivacaftor component of Trikafta and its major metabolites). This finding has not been observed in older animals. The potential relevance of these findings in humans is unknown.
Effects on laboratory tests.
See Section 4.4 Special Warnings and Precautions for Use, Elevated transaminases and hepatic injury.
Patients after organ transplantation.
Trikafta has not been studied in patients with CF who have undergone organ transplantation.
Therefore, use in transplanted patients is not recommended (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions for interactions with ciclosporin, everolimus, sirolimus or tacrolimus).
Use in the elderly.
Clinical trials of Trikafta did not include a sufficient number of patients aged 65 years and older to determine whether they respond differently from younger patients.
Paediatric use.
The safety and efficacy of Trikafta in children aged less than 2 years have not been established (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties).4.5 Interactions with Other Medicines and Other Forms of Interactions
Medicinal products affecting the pharmacokinetics of Trikafta.
CYP3A inducers.
Elexacaftor, tezacaftor and ivacaftor are substrates of CYP3A (ivacaftor is a sensitive substrate of CYP3A). Concomitant use of CYP3A inducers may result in reduced exposures and thus reduced Trikafta efficacy. Co-administration of ivacaftor with rifampicin, a strong CYP3A inducer, significantly decreased ivacaftor area under the curve (AUC) by 89%. Elexacaftor and tezacaftor exposures are expected to decrease during co-administration with strong CYP3A inducers; therefore, co-administration of Trikafta with strong CYP3A inducers is not recommended (see Section 4.4 Special Warnings and Precautions for Use).
Examples of strong CYP3A inducers include: rifampicin, rifabutin, phenobarbital, carbamazepine, phenytoin, and St. John's wort (Hypericum perforatum).
CYP3A inhibitors.
Co-administration with itraconazole, a strong CYP3A inhibitor, increased elexacaftor AUC by 2.8-fold and tezacaftor AUC by 4.0- to 4.5-fold. When co-administered with itraconazole and ketoconazole, ivacaftor AUC increased by 15.6-fold and 8.5-fold, respectively. The dose of Trikafta should be reduced when co-administered with strong CYP3A inhibitors (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration, Table 3).
Examples of strong CYP3A inhibitors include: ketoconazole, itraconazole, posaconazole, and voriconazole;
telithromycin and clarithromycin.
Simulations indicated that co-administration with moderate CYP3A inhibitors may increase elexacaftor and tezacaftor AUC by approximately 1.9- to 2.3-fold. Co-administration of fluconazole increased ivacaftor AUC by 2.9-fold. The dose of Trikafta should be reduced when co-administered with moderate CYP3A inhibitors (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration, Table 3).
Examples of moderate CYP3A inhibitors include: fluconazole, erythromycin, verapamil.
Co-administration of Trikafta with grapefruit juice, which contains one or more components that moderately inhibit CYP3A, may increase exposure of elexacaftor, tezacaftor and ivacaftor. Food or drink containing grapefruit should be avoided during treatment with Trikafta (see Section 4.2 Dose and Method of Administration).
The effects of co-administered drugs on the exposure of elexacaftor, tezacaftor and/or ivacaftor are shown in Table 4 (see Section 4.2 Dose and Method of Administration).

Medicinal products affected by Trikafta.
CYP2C9 substrates.
Ivacaftor may inhibit CYP2C9; therefore, monitoring of the international normalised ratio (INR) during co-administration of Trikafta with warfarin is recommended. Other medicinal products for which exposure may be increased by Trikafta include glimepiride and glipizide; these medicinal products should be used with caution.
Potential for interaction with transporters.
Co-administration of ivacaftor or tezacaftor/ivacaftor with digoxin, a sensitive P-glycoprotein (P-gp) substrate, increased digoxin AUC by 1.3-fold, consistent with weak inhibition of P-gp by ivacaftor. Administration of Trikafta may increase systemic exposure of medicinal products that are sensitive substrates of P-gp, which may increase or prolong their therapeutic effect and adverse reactions. When used concomitantly with digoxin or other substrates of P-gp with a narrow therapeutic index such as ciclosporin, everolimus, sirolimus, and tacrolimus, caution and appropriate monitoring should be used.
Elexacaftor and M23-ELX (active metabolite) inhibit uptake by OATP1B1 and OATP1B3 in vitro. Tezacaftor/ivacaftor increased the AUC of pitavastatin, an OATP1B1 substrate, by 1.2-fold. Coadministration of Trikafta may increase exposures of medicinal products that are substrates of these transporters, such as statins, glyburide, nateglinide and repaglinide. When used concomitantly with substrates of OATP1B1 or OATP1B3, caution and appropriate monitoring should be used. Bilirubin is an OATP1B1 and OATP1B3 substrate. In Study 445-102, mild increases in mean total bilirubin were observed (up to 4.0 micromol/L change from baseline). This finding is consistent with the in vitro inhibition of bilirubin transporters OATP1B1 and OATP1B3 by elexacaftor and M23-ELX.
Hormonal contraceptives.
Trikafta has been studied with ethinyl estradiol/levonorgestrel and was found to have no clinically relevant effect on the exposures of the oral contraceptive. Trikafta is not expected to have an impact on the efficacy of oral contraceptives.
The effects of elexacaftor, tezacaftor and/or ivacaftor on the exposure of co-administered drugs are shown in Table 5.

4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data available on the effect of elexacaftor, tezacaftor, and ivacaftor on fertility in humans.
Elexacaftor impaired male and female fertility in rats at oral doses of 75 mg/kg/day and 35 mg/kg/day in the respective sexes (yielding systemic exposure in animals approximately 6 and 7 times greater, respectively, than that in patients at the MRHD based on summed AUCs of the elexacaftor component of Trikafta and its major active metabolite, M23-ELX).
Tezacaftor did not affect fertility or reproductive performance indices in male and female rats at oral doses up to 100 mg/kg/day (yielding systemic exposure in animals approximately 3 times greater than that in patients at the MRHD based on summed AUCs of the tezacaftor component of Trikafta and its pharmacologically active metabolite, M1-TEZ).
Ivacaftor impaired fertility and reproductive performance indices in male and female rats at an oral dose of 200 mg/kg/day (yielding systemic exposure in animals approximately 10 and 5 times greater, respectively, than that in patients at the MRHD based on summed AUCs of the ivacaftor component of Trikafta and its major metabolites) when dams were dosed prior to and during early pregnancy. The pregnancy rate was decreased, oestrus cycling was disrupted, and pre-implantation loss was increased. These effects occurred in the presence of significant maternal toxicity. No effects on male or female fertility and reproductive performance indices were observed at ≤ 100 mg/kg/day (yielding systemic exposure in animals approximately 5 and 3 times greater, respectively, than that in patients at the MRHD based on the summed AUCs of the ivacaftor component of Trikafta and its major metabolites).
(Category B3)
Category B3 drugs have been taken by only a limited number of pregnant women and women of childbearing age, without an increase in the frequency of malformation or other direct or indirect harmful effects on the human fetus having been observed.
Studies in animals have shown evidence of an increased occurrence of fetal damage, the significance of which is considered uncertain in humans.
Elexacaftor, tezacaftor, ivacaftor and/or their metabolites were shown to cross the placenta in laboratory animal species (rats and/or rabbits).
Elexacaftor.
Elexacaftor was not teratogenic in rats at oral doses up to 40 mg/kg/day or up to 125 mg/kg/day in rabbits (yielding systemic exposure in animals approximately 9 and 4 times greater, respectively, than that in patients at the MRHD based on summed AUCs of the elexacaftor component of Trikafta and M23-ELX [for rat], or AUC of the elexacaftor component of Trikafta [for rabbit]). Effects on embryofetal development were limited to lower mean fetal body weight (at ≥ 25 mg/kg/day). Pup birth and postnatal body weights were reduced in rats with maternal treatment at 10 mg/kg/day during gestation and lactation.
Tezacaftor.
No evidence of harm to the fetus was observed with tezacaftor in developmental toxicity study in rats at oral doses up to 100 mg/kg/day (yielding systemic exposure in animals approximately 3 times greater than that in patients at the MRHD based on summed AUCs of the tezacaftor component of Trikafta and its pharmacologically active M1 metabolite, M1-TEZ). In the rabbit, lower fetal body weights were noted at an oral dose of 50 mg/kg/day (the highest dose tested; yielding exposure around the same as at the MRHD), which occurred in conjunction with significant maternal toxicity. However, no effects on embryo fetal survival and no malformations were observed with tezacaftor in the species. Fetal body weight was unaffected in rabbits at 25 mg/kg/day (yielding exposure 4 times lower than that at the MRHD based on summed AUCs of tezacaftor and its M1 metabolite).
Ivacaftor.
Developmental toxicity studies with ivacaftor revealed no teratogenicity in rats at oral doses up to 200 mg/kg/day or rabbits at oral doses up to 100 mg/kg/day (yielding systemic exposure in the respective animal species approximately 5 and ≥ 3 times greater, than that in patients at the MRHD based on summed AUCs of the ivacaftor component of Trikafta and its major metabolites). Fetal weight was decreased and the incidence of minor fetal skeletal abnormalities was increased in rats treated at 200 mg/kg/day; these effects were observed in conjunction with maternal toxicity.
No adequate and well-controlled studies of Trikafta in pregnant women have been conducted. Because animal reproduction studies are not always predictive of human response, Trikafta should be used during pregnancy only if the potential benefits outweigh the potential risks.
Elexacaftor, tezacaftor and ivacaftor are excreted into the milk of lactating female rats. Exposure of 14C-elexacaftor, 14C-tezacaftor and 14C-ivacaftor in milk was approximately 0.4, 2, and 1.5 times, respectively, the value observed in plasma (based on AUC0-24h). Because it is not known if elexacaftor, tezacaftor, ivacaftor, or their metabolites are excreted in human milk, Trikafta should be used during breastfeeding only if the potential benefit outweighs the potential risks to the infant.4.7 Effects on Ability to Drive and Use Machines
Trikafta is not expected to have an impact on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
The safety profile of Trikafta is based on data from 510 patients in two double-blind, controlled, phase 3 studies of 24 weeks and 4 weeks treatment duration (Studies 445-102 and 445-103). In the two controlled phase 3 studies, a total of 257 patients aged 12 years and older received at least one dose of Trikafta.
In Study 445-102, the proportion of patients who discontinued study drug prematurely due to adverse events was 1% for Trikafta-treated patients and 0% for placebo-treated patients.
Serious adverse drug reactions that occurred more frequently in Trikafta-treated patients compared to placebo were rash events in 3 (1.5%) Trikafta-treated patients vs.1 (0.5%) placebo. The most common (≥ 10%) adverse drug reactions in patients treated with Trikafta were headache, diarrhoea and upper respiratory tract infection.
The safety profile of Trikafta was generally similar across all subgroups of patients, including analysis by age, sex, baseline percent predicted FEV1 (ppFEV1), and geographic regions.
Table 6 shows adverse events with an incidence of at least 10% in any treatment group from the double-blind, placebo-controlled, phase 3 clinical Study 445-102 (24 weeks duration).

Tabulated list of adverse reactions.
Table 7 shows adverse drug events occurring in ≥ 8% of Trikafta-treated patients and at a frequency higher than placebo by ≥ 1% in Study 445-102. Adverse drug events for Trikafta are ranked under the MedDRA frequency classification: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).
 Safety data from the following studies were consistent with the safety data observed in Study 445-102.
Safety data from the following studies were consistent with the safety data observed in Study 445-102.
A 4-week, randomised, double-blind, active-controlled study in 107 patients (Study 445-103).
A 192-week, open-label safety and efficacy study (Study 445-105) for patients rolled over from Studies 445-102 and 445-103.
An 8-week, randomised, double-blind, active-controlled study in 258 patients (Study 445-104).
A 24-week, open-label study (Study 445-106) in 66 patients aged 6 to less than 12 years.
A 24-week, open-label study (Study 445-111) in 75 patients aged 2 to less than 6 years.
A 192-week, two-part (part A and part B), open-label safety and efficacy study (Study 445-107) in patients aged 6 years and older who rolled over from Study 445-106, with part A analysis (96 weeks) performed on 64 patients.
A 24-week, randomised, double-blind, placebo-controlled study (Study 445-124) in 307 patients aged 6 years and older.
Detailed description of selected adverse events.
Laboratory abnormalities. Transaminase elevations.
In Study 445-102, the incidence of maximum transaminase (ALT or AST) > 8, > 5, or > 3 x the ULN was 1.5%, 2.5%, and 7.9% in Trikafta-treated patients and 1.0%, 1.5%, and 5.5% in placebo-treated patients. The incidence of adverse reactions of transaminase elevations was 10.9% in Trikafta-treated patients and 4.0% in placebo-treated patients. No Trikafta-treated patients discontinued treatment for elevated transaminases (see Section 4.4 Special Warnings and Precautions for Use).
During Study 445-106 in patients aged 6 to less than 12 years, the incidence of maximum transaminase (ALT or AST) > 8, > 5, and > 3 x ULN were 0%, 1.5%, and 10.6%, respectively. No Trikafta-treated patients had transaminase elevation > 3 x ULN associated with elevated total bilirubin > 2 x ULN or discontinued treatment due to transaminase elevations (see Section 4.4 Special Warnings and Precautions for Use).
During Study 445-111 in patients aged 2 to less than 6 years, the incidence of maximum transaminase (ALT or AST) > 8, > 5, and > 3 x ULN were 1.3%, 2.7%, and 8.0%, respectively. No Trikafta-treated patients had transaminase elevation > 3 x ULN associated with elevated total bilirubin > 2 x ULN or discontinued treatment due to transaminase elevations (see Section 4.4 Special Warnings and Precautions for Use).
Rash events.
Studies in Trikafta-treated patients above 12 years of age showed an incidence of rash events (e.g. rash, rash pruritic) of 10.9% (study 445-102) compared to 6.5% in placebo-treated patients. The paediatric population showed a higher incidence rate (see section Paediatric population for further details). The incidence of rash events by patient sex was 5.8% in males and 16.3% in females in Trikafta-treated patients and 4.8% in males and 8.3% in females in placebo-treated patients. In patients treated with Trikafta, the incidence of rash events was 20.5% in females taking hormonal contraceptive and 13.6% in females not taking hormonal contraceptive (see Section 4.4 Special Warnings and Precautions for Use).
Overall, rash events typically occur during the first month of therapy. Most events were mild to moderate in severity, and in rare cases, rash was associated with additional symptoms such as fever or facial swelling. In the majority of cases, administration of Trikafta was continued and the rash resolved without treatment.
A role for hormonal contraceptives in the occurrence of rash cannot be excluded. For patients taking hormonal contraceptives who develop rash, consider interrupting Trikafta and hormonal contraceptives. Following the resolution of rash, consider resuming Trikafta without the hormonal contraceptives. If rash does not recur, resumption of hormonal contraceptives can be considered.
Paediatric population. Rash.
While studies in patients above 12 years of age showed an incidence rate of 10.9% (study 445-102), patients between 6 and 11 years of age had an incidence rate of 24.2% (study 445-106). During study 445-111 in patients aged 2 to less than 6 years, 15 (20.0%) subjects had at least 1 rash event, 4 (9.8%) females and 11 (32.4%) males.
Increased creatine phosphokinase.
In Study 445-102, the incidence of maximum creatine phosphokinase > 5 x the ULN was 10.4% in Trikafta-treated patients and 5.0% in placebo-treated patients. No Trikafta-treated patients discontinued treatment for increased creatine phosphokinase.
In Study 445-111 CK elevation occurred in 1 (1.3%) subject. The mean (SD) increase in CK ranged from 25.7 (82.4) U/L at Day 15 to 41.7 (47.3) U/L at Week 20. The majority of subjects had CK levels ≤ 2 x ULN; no subjects had CK levels > 5 x ULN.
Increased blood pressure.
In Study 445-102, the maximum increase from baseline in mean systolic and diastolic blood pressure was 3.5 mmHg and 1.9 mmHg, respectively, for Trikafta-treated patients (baseline: 113 mmHg systolic and 69 mmHg diastolic) and 0.9 mmHg and 0.5 mmHg, respectively for placebo-treated patients (baseline: 114 mmHg systolic and 70 mmHg diastolic).
The proportion of patients who had systolic blood pressure > 140 mmHg or diastolic blood pressure > 90 mmHg on at least two occasions was 5.0% and 3.0% in Trikafta-treated patients, respectively, compared with 3.5% and 3.5% in placebo-treated patients, respectively.
Post-marketing experience.
The following adverse reactions have been identified during post approval use of Trikafta. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Liver failure leading to transplantation in patients with and without pre-existing advanced liver disease (e.g. cirrhosis, portal hypertension). Liver injury characterised by concomitant transaminase (ALT and AST) and total bilirubin elevations (see Section 4.4 Special Warnings and Precautions for Use).
Immune system disorders.
Anaphylaxis, hypersensitivity.
Reporting suspected adverse effects.
Reporting of suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
No specific antidote is available for overdose with Trikafta. Treatment of overdose consists of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Respiratory system, other respiratory system products; ATC code: R07AX32.
Mechanism of action.
Elexacaftor and tezacaftor are CFTR correctors that bind to different sites on the CFTR protein and have an additive effect in facilitating the cellular processing and trafficking of select mutant forms of CFTR (including F508del CFTR) to increase the amount of CFTR protein delivered to the cell surface compared to either molecule alone. Ivacaftor potentiates the channel open probability (or gating) of the CFTR protein at the cell surface.
The combined effect of elexacaftor, tezacaftor and ivacaftor is increased quantity and function of CFTR at the cell surface, resulting in increased CFTR activity as measured by CFTR mediated chloride transport. (See Clinical efficacy).
CFTR chloride transport assay in Fischer rat thyroid (FRT) cells expressing mutant CFTR.
The chloride transport response of mutant CFTR protein to ELX/TEZ/IVA was determined in Ussing chamber electrophysiology studies using a panel of FRT cell lines transfected with individual CFTR mutations. ELX/TEZ/IVA increased chloride transport in FRT cells expressing select CFTR mutations.
The in vitro CFTR chloride transport response threshold was designated as a net increase of at least 10% of normal over baseline because it is predictive or reasonably expected to predict clinical benefit. For individual mutations, the magnitude of the net change over baseline in CFTR-mediated chloride transport in vitro is not correlated with the magnitude of clinical response.
Clinical outcomes were consistent with in vitro results and indicate that a single elexacaftor/tezacaftor/ivacaftor responsive mutation is sufficient to result in a significant clinical response (see Clinical efficacy).
Table 8 lists responsive CFTR mutations based on clinical response and/or in vitro data in FRT cells indicating that elexacaftor/tezacaftor/ivacaftor increases chloride transport to at least 10% of normal over baseline.

Clinical trials.
Pharmacodynamic effects. Effects on sweat chloride.
In Study 445-102 (patients with an F508del mutation on one allele and a mutation on the second allele that results in either no CFTR protein or a CFTR protein that is not responsive to ivacaftor and tezacaftor/ivacaftor (minimal function mutation)), a reduction in sweat chloride was observed from baseline at Week 4 and sustained through the 24-week treatment period.
The treatment difference of Trikafta compared to placebo for mean absolute change in sweat chloride from baseline through Week 24 was -41.8 mmol/L (95% CI: -44.4, -39.3; P < 0.0001).
In Study 445-103 (patients homozygous for the F508del mutation), the treatment difference of Trikafta compared to tezacaftor/ivacaftor for mean absolute change in sweat chloride from baseline at Week 4 was -45.1 mmol/L (95% CI: -50.1, -40.1; P < 0.0001).
In Study 445-104 (patients heterozygous for the F508del mutation and a gating or residual function mutation on the second allele), following a 4-week ivacaftor or tezacaftor/ivacaftor run-in period, the mean absolute change in sweat chloride from baseline through Week 8 for the Trikafta group was -22.3 mmol/L (95% CI: -24.5, -20.2; P < 0.0001). The treatment difference of Trikafta compared to the control group (ivacaftor or tezacaftor/ivacaftor) was -23.1 mmol/L (95% CI: -26.1, -20.1; P < 0.0001).
In Study 445-106 (patients aged 6 to less than 12 years who are homozygous for the F508del mutation or heterozygous for the F508del mutation and a minimal function mutation), the mean absolute change in sweat chloride from baseline through Week 24 was -60.9 mmol/L (95% CI: -63.7, -58.2).
In Study 445-111 (patients aged 2 to less than 6 years who are homozygous for the F508del mutation or heterozygous for the F508del mutation and a minimal function mutation), the mean absolute change in sweat chloride from baseline through Week 24 was -57.9 mmol/L (95% CI: -61.3, -54.6).
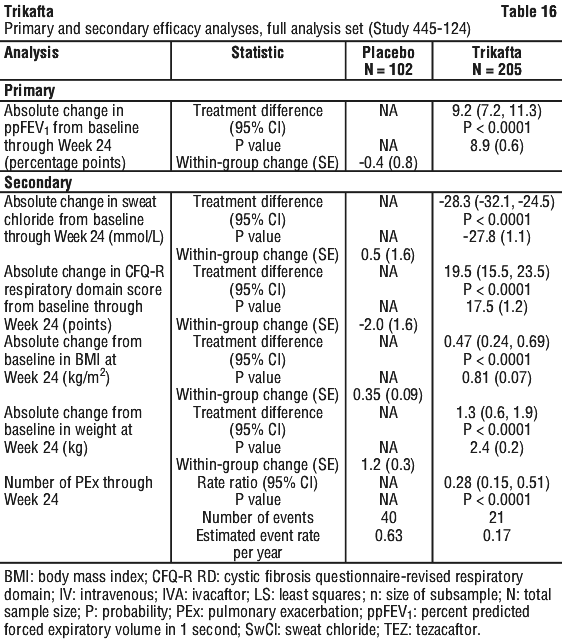
In Study 445-124 (patients aged 6 years and older with a qualifying non-F508del, ELX/TEZ/IVA-responsive mutation [see Table 15]), the mean absolute change in sweat chloride from baseline through Week 24 compared to placebo was -28.3 mmol/L (95% CI: -32.1, -24.5 mmol/L; P < 0.0001).
Cardiovascular effects. Effect on QT interval.
At doses up to 2 times the maximum recommended dose of elexacaftor and 3 times the maximum recommended dose of tezacaftor and ivacaftor, the QT/QTc interval in healthy subjects was not prolonged to any clinically relevant extent.
Heart rate.
In Study 445-102, mean decreases in heart rate of 3.7 to 5.8 beats per minute (bpm) from baseline (76 bpm) were observed in Trikafta-treated patients.
Clinical efficacy. The efficacy of Trikafta in patients with CF was demonstrated in four phase 3, double-blind, controlled studies (Studies 445-102, 445-103, 445-104 and 445-124), a phase 3 open-label extension study (Study 445-105), and two phase 3 open-label studies (Study 445-106 and Study 445-111). These studies enrolled CF patients with at least one F508del mutation or a mutation responsive to Trikafta listed in Table 8. Significant clinical benefit was demonstrated in all studies.
Patients in Studies 445-102, 445-103, 445-104, 445-106, 445-111 and 445-124 continued on their CF therapies (e.g. bronchodilators, inhaled antibiotics, dornase alfa, and hypertonic saline), but discontinued any previous CFTR modulator therapies, except for study drugs. Patients had a confirmed diagnosis of CF and at least one F508del mutation or a mutation responsive based on clinical or in vitro evidence.
Patients in Studies 445-102, 445-103, 445-104, 445-106, 445-111 and 445-124 who had lung infection with organisms associated with a more rapid decline in pulmonary status, including but not limited to Burkholderia cenocepacia, Burkholderia dolosa, or Mycobacterium abscessus, or who had an abnormal liver function test at screening (ALT, AST, ALP, or GGT ≥ 3 x ULN, or total bilirubin ≥ 2 x ULN), were excluded. In Study 445-111, patients who had ALT or AST ≥ 2 x ULN were also excluded.
Patients in Studies 445-102 and 445-103 were eligible to roll over into a 192-week open label extension study (Study 445-105). Patients in Studies 445-104, 445-106, 445-111 and 445-124 were eligible to roll over into distinct open label extension studies.
Study 445-102: study in patients who had an F508del mutation on one allele and a mutation on the second allele that results in either no CFTR/ non-responsive CFTR protein.
Study 445-102 was a 24-week, randomised, double-blind, placebo-controlled study in patients who had an F508del mutation on one allele and a minimal function mutation on the second allele.* A total of 403 patients aged 12 years and older (mean age 26.2 years) were randomised and dosed to receive Trikafta or placebo. Patients had a ppFEV1 at screening between 40-90%. The mean ppFEV1 at baseline was 61.4% (range: 32.3%, 97.1%).
* Contact sponsor (see Section 8 Sponsor) for list of mutations enrolled in Study 445-102.
In Study 445-102 the primary endpoint was mean absolute change in ppFEV1 from baseline through Week 24. Treatment with Trikafta compared to placebo resulted in statistically significant improvement in ppFEV1 of 14.3 percentage points (95% CI: 12.7, 15.8; P < 0.0001) (see Table 9). Mean improvement in ppFEV1 was rapid in onset (Day 15) and sustained through the 24-week treatment period (see Figure 1). Improvements in ppFEV1 were observed regardless of age, baseline ppFEV1, sex, and geographic region. A total of 18 patients receiving Trikafta had ppFEV1 < 40 at baseline. The safety and efficacy in this subgroup were comparable to those observed in the overall population. See Table 9 for a summary of primary and key secondary outcomes.
 At Week 24 the proportion of patients who remained free from pulmonary exacerbations was significantly higher for patients treated with Trikafta compared with placebo. The rate ratio of exacerbations through Week 24 in patients treated with Trikafta was 0.37 (95% CI: 0.25, 0.55; P < 0.0001), representing a reduction relative to placebo of 63% (see Figure 2).
At Week 24 the proportion of patients who remained free from pulmonary exacerbations was significantly higher for patients treated with Trikafta compared with placebo. The rate ratio of exacerbations through Week 24 in patients treated with Trikafta was 0.37 (95% CI: 0.25, 0.55; P < 0.0001), representing a reduction relative to placebo of 63% (see Figure 2).


Study 445-103: study in patients who are homozygous for the F508del mutation and randomised to Trikafta or Symdeko tablets.
Study 445-103 was a 4-week, randomised, double-blind, active-controlled study in patients who are homozygous for the F508del mutation. A total of 107 patients aged 12 years and older (mean age 28.4 years) received Symdeko (tezacaftor/ivacaftor and ivacaftor regimen) during a 4-week open-label run-in period and were then randomised and dosed to receive Trikafta or Symdeko during a 4-week double-blind treatment period. Patients had a ppFEV1 at screening between 40-90%. The mean ppFEV1 at baseline, following the Symdeko run-in period was 60.9% (range: 35.0%, 89.0%).
In Study 445-103 the primary endpoint was mean absolute change in ppFEV1 from baseline at Week 4 of the double-blind treatment period. Treatment with Trikafta compared to the Symdeko resulted in a statistically significant improvement in ppFEV1 of 10.0 percentage points (95% CI: 7.4, 12.6; P < 0.0001) (see Table 10). Improvements in ppFEV1 were observed regardless of age, sex, baseline ppFEV1, and geographic region. See Table 10 for a summary of primary and key secondary outcomes.
 See Figure 3.
See Figure 3.

Study 445-104: study in patients aged 12 years and older who are heterozygous for the F508del mutation and a gating or residual function mutation.
Study 445-104 was an 8-week, randomised, double-blind, active-controlled study in patients who were heterozygous for the F508del mutation and a gating or residual function (RF) mutation on the second allele. A total of 258 patients aged 12 years and older received either Kalydeco (for F/G patients) or Symdeko (for F/RF patients) during a 4-week open label run in period and were dosed during the treatment period. Patients with the F/R117H genotype received ivacaftor during the run-in period. The mean age at baseline, following the run-in period was 37.7 years. Patients were then randomised to the Trikafta group or remained on the CFTR modulator therapy received during the run-in period. Patients had a ppFEV1 screening between 40-90%. The mean ppFEV1 at baseline was 67.6% (range: 29.7%, 113.5%).
Following a 4-week Kalydeco or Symdeko run-in period, the primary endpoint of within group mean absolute change in ppFEV1 from baseline through Week 8 for the Trikafta group resulted in statistically significant improvement in ppFEV1 of 3.7 percentage points (95% CI: 2.8, 4.6; P < 0.0001) (see Table 11). Mean improvement in ppFEV1 was observed at the first assessment on Day 15. Overall improvements in ppFEV1 were observed regardless of age, sex, baseline ppFEV1 geographic region, and genotype groups (F/G or F/RF).
See Table 11 for a summary of primary and secondary outcomes in the overall trial population.
In a subgroup analysis of patients with an F/G genotype, the treatment difference of Trikafta (N=50) compared with Kalydeco (N=45) for mean absolute change in ppFEV1 was 5.8 percentage points (95% CI: 3.5, 8.0). In a subgroup analysis of patients with an F/RF genotype, the treatment difference of Trikafta (N=82) compared with Symdeko (N=81) for mean absolute change in ppFEV1 was 2.0 percentage points (95% CI: 0.5, 3.4). The results of the F/G and the F/RF genotype subgroups for improvement in sweat chloride and CFQ-R respiratory domain score were consistent with the overall results.

Study 445-105: a 192 week open-label study in patients aged 12 years and older rolled over from studies 445‑102 and 445-103.
Study 445-105 was a 192-week open-label extension study to evaluate the safety and efficacy of long-term treatment with Trikafta conducted in patients who rolled over from Studies 445-102 (N = 400) and 445-103 (N = 107). In this open-label extension study, all patients received Trikafta for the duration of the study.
In Study 445-105, patients from the control arms in the parent studies showed improvements in efficacy endpoints consistent with those observed in subjects who received Trikafta in the parent studies. Patients from the control arms as well as patients who received Trikafta in the parent studies showed sustained improvements in ppFEV1 (see Figure 4 and Figure 5) and other efficacy endpoints (see Table 12).



Study 445-106: study in patients aged 6 to less than 12 years old who are homozygous for the F508del mutation or heterozygous for the F508del mutation and a minimal function mutation.
Study 445-106 was a 24-week open-label study in patients who were homozygous for the F508del mutation or heterozygous for the F508del mutation and a minimal function mutation. A total of 66 patients aged 6 to less than 12 years (mean age at baseline 9.3 years) were dosed according to weight. Patients weighing < 30 kg at baseline were administered elexacaftor 100 mg once daily (qd)/tezacaftor 50 mg qd/ivacaftor 75 mg every 12 hours (q12h), and patients weighing ≥ 30 kg at baseline were administered elexacaftor 200 mg qd/tezacaftor 100 mg qd/ivacaftor 150 mg q12h. Patients had a ppFEV1 ≥ 40% and weighed ≥ 15 kg at screening. The mean ppFEV1 at baseline was 88.8% (range: 39.0%, 127.1%).
The pharmacokinetic profile, safety, and efficacy of Trikafta in patients with CF aged 6 to less than 12 years are supported by evidence from studies of Trikafta in patients aged 12 years and older (Studies 445-102, 445-103, and 445-104), with additional data from a 24-week, open-label, phase 3 study in 66 patients aged 6 to less than 12 years (Study 445-106).
In Study 445-106 the primary endpoint of safety and tolerability was evaluated through 24 weeks. Secondary endpoints were evaluation of pharmacokinetics, and efficacy including absolute change in ppFEV1, sweat chloride (see Section 5.1 Pharmacodynamic Properties), CFQ-R respiratory domain score, and LCI2.5 from baseline through Week 24; measure of growth parameters (weight, height, BMI; and associated z-scores) from baseline at Week 24; and number of pulmonary exacerbations from baseline through Week 24. See Table 13 for a summary of secondary efficacy outcomes.

Study 445-107: an ongoing open label study to evaluate safety and efficacy in patients aged 6 to 11 years who completed study 445-106.
A 192-week, two-part (part A and part B), open-label extension study to evaluate the safety and efficacy of long-term treatment with ELX/TEZ/IVA is being conducted in patients who completed Study 445-106. Part A (96 weeks) analysis was conducted in 64 paediatric patients aged 6 years and older and showed sustained improvements in ppFEV1, SwCl, CFQ-R RD score, and LCI2.5, consistent with the results observed in the Study 445-106. Secondary efficacy endpoints of the interim analysis are summarised in Table 14.

Study 445-111: study in patients aged 2 to less than 6 years old who had at least one F508del mutation or a mutation known to be responsive to Trikafta.
Study 445-111 was a 24-week, open-label study in patients aged 2 to less than 6 years (mean age at baseline 4.1 years). Patients who had at least one F508del mutation or a mutation known to be responsive to Trikafta were eligible for the study. A total of 75 patients who were homozygous for the F508del mutation or heterozygous for the F508del mutation and a minimal function mutation were enrolled and dosed according to weight. Patients weighing 10 kg to < 14 kg at baseline were administered ELX 80 mg once daily (qd)/TEZ 40 mg qd/IVA 60 mg once every morning and IVA 59.5 mg once every evening. Patients weighing ≥ 14 kg at baseline were administered ELX 100 mg qd/TEZ 50 mg qd/IVA 75 mg q12h.
The pharmacokinetic profile, safety, and efficacy of Trikafta in patients with CF aged 2 to less than 6 years are supported by evidence from studies of Trikafta in patients aged 12 years and older (Studies 445-102, 445-103 and 445-104), with additional data from a 24-week, open-label, phase 3 study in 75 patients aged 2 to less than 6 years (Study 445-111).
In Study 445-111 the primary endpoint of safety and tolerability was evaluated through 24 weeks. Secondary endpoints were an evaluation of pharmacokinetics, and efficacy endpoints of absolute change in sweat chloride (see Section 5.1 Pharmacodynamic Properties) and the mean absolute change in LCI2.5 from baseline through Week 24 assessed only on patients aged 3 years and older at screening was 0.83 (95% CI: -1.01, -0.66).
Study 445-124: study in patients aged 6 years and over with at least one qualifying non F508del, elexacaftor/tezacaftor/ivacaftor-responsive mutation.
Study 445-124 was a 24 week, randomised, placebo-controlled, double-blind, parallel group study evaluating safety and efficacy of Trikafta in patients with CF aged 6 years and older without an F508del mutation. Patients who had at least one qualifying non-F508del, elexacaftor/tezacaftor/ivacaftor-responsive mutation (see Table 15) and did not have an exclusionary (other elexacaftor/tezacaftor/ivacaftor - responsive) mutation were eligible for the study. A total of 307 patients were enrolled and dosed according to age and weight. Patients ≥ 6 to < 12 years weighing < 30 kg at baseline were administered elexacaftor 100 mg qd/ tezacaftor 50 mg qd/ ivacaftor 75 mg q12h. Patients ≥ 6 to < 12 years weighing ≥ 30 kg at baseline were administered elexacaftor 200 mg qd/ tezacaftor 100 mg qd/ ivacaftor 150 mg q12h. Patients ≥ 12 years at baseline were administered elexacaftor 200 mg qd/ tezacaftor 100 mg qd/ ivacaftor 150 mg q12h. Patients had a ppFEV1 ≥ 40% and ≤ 100% and aged 6 years or older at screening. The mean ppFEV1 at baseline was 67.7% (range: 34.0%, 108.7%)]. See Table 15.
 In Study 445-124, the primary endpoint of efficacy was ppFEV1. Secondary endpoints were absolute change in sweat chloride, CFQ-R respiratory domain score, growth parameters (BMI, weight), and number of PEx. See Table 16 for a summary of primary and secondary efficacy outcomes.
In Study 445-124, the primary endpoint of efficacy was ppFEV1. Secondary endpoints were absolute change in sweat chloride, CFQ-R respiratory domain score, growth parameters (BMI, weight), and number of PEx. See Table 16 for a summary of primary and secondary efficacy outcomes.
5.2 Pharmacokinetic Properties
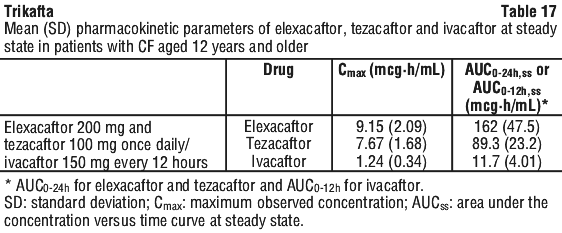
The pharmacokinetics of elexacaftor, tezacaftor and ivacaftor are similar between healthy adult subjects and patients with CF. Following initiation of once-daily dosing of elexacaftor and tezacaftor and twice-daily dosing of ivacaftor, plasma concentrations of elexacaftor, tezacaftor and ivacaftor reach steady state within approximately 7 days for elexacaftor, within 8 days for tezacaftor, and within 3-5 days for ivacaftor. Upon dosing elexacaftor/tezacaftor/ivacaftor to steady state, the accumulation ratio is approximately 3.6 for elexacaftor, 2.8 for tezacaftor and 4.7 for ivacaftor. Key pharmacokinetic parameters for elexacaftor, tezacaftor and ivacaftor at steady state in patients with CF aged 12 years and older are shown in Table 17.
Absorption.
The absolute bioavailability of elexacaftor when administered orally in the fed state is approximately 80%. Elexacaftor is absorbed with a median (range) time to maximum concentration (tmax) of approximately 6 hours (4 to 12 hours) while the median (range) tmax of tezacaftor and ivacaftor is approximately 3 hours (2 to 4 hours) and 4 hours (3 to 6 hours), respectively.
Elexacaftor exposure (AUC) increases approximately 1.9- to 2.5-fold when administered with a moderate-fat meal relative to fasted conditions. Ivacaftor exposure increases approximately 2.5- to 4.0-fold when administered with fat-containing meals relative to fasted conditions, while food has no effect on the exposure of tezacaftor (see Section 4.2 Dose and Method of Administration).
Distribution.
Elexacaftor is > 99% bound to plasma proteins and tezacaftor is approximately 99% bound to plasma proteins, in both cases primarily to albumin. Ivacaftor is approximately 99% bound to plasma proteins, primarily to albumin, and also to alpha 1-acid glycoprotein and human gamma-globulin. After oral administration of Trikafta, the mean (± SD) apparent volume of distribution of elexacaftor, tezacaftor and ivacaftor was 53.7 L (17.7), 82.0 L (22.3) and 293 L (89.8), respectively. Elexacaftor, tezacaftor and ivacaftor do not partition preferentially into human red blood cells.
Metabolism.
Elexacaftor is metabolised extensively in humans, mainly by CYP3A4/5. Following oral administration of a single dose of 200 mg 14C-elexacaftor to healthy male subjects, M23-ELX was the only major circulating metabolite. M23-ELX is considered pharmacologically active.
Tezacaftor is metabolised extensively in humans, mainly by CYP3A4/5. Following oral administration of a single dose of 100 mg 14C-tezacaftor to healthy male subjects, M1-TEZ, M2-TEZ, and M5-TEZ were the 3 major circulating metabolites of tezacaftor in humans. M1-TEZ has similar apparent potency to that of tezacaftor and is considered pharmacologically active. M2-TEZ is much less pharmacologically active than tezacaftor or M1-TEZ, and M5-TEZ is not considered pharmacologically active. Another minor circulating metabolite, M3-TEZ, is formed by direct glucuronidation of tezacaftor.
Ivacaftor is also metabolised extensively in humans. In vitro and in vivo data indicate that ivacaftor is metabolised primarily by CYP3A4/5. M1-IVA and M6-IVA are the two major metabolites of ivacaftor in humans. M1-IVA has approximately one-sixth the potency of ivacaftor and is considered pharmacologically active. M6-IVA is not considered pharmacologically active.
Excretion.
Following multiple dosing in the fed state, the mean (± SD) apparent clearance values of elexacaftor, tezacaftor and ivacaftor at steady state were 1.18 (0.29) L/h, 0.79 (0.10) L/h and 10.2 (3.13) L/h, respectively. The mean (SD) terminal half-lives of elexacaftor, tezacaftor and ivacaftor following administration of the elexacaftor/tezacaftor/ivacaftor fixed dose combination tablets are approximately 24.7 (4.87) hours, 60.3 (15.7) hours and 13.1 (2.98) hours, respectively. The mean (SD) effective half-lives of elexacaftor, tezacaftor and ivacaftor following administration of the elexacaftor/tezacaftor/ivacaftor fixed dose combination tablets are approximately 27.4 (9.31) hours, 25.1 (4.93) hours and 15.0 (3.92) hours, respectively.
Following oral administration of 14C-elexacaftor alone, the majority of elexacaftor (87.3%) was eliminated in the faeces, primarily as metabolites.
Following oral administration of 14C-tezacaftor alone, the majority of the dose (72%) was excreted in the faeces (unchanged or as the M2-TEZ) and about 14% was recovered in urine (mostly as M2-TEZ), resulting in a mean overall recovery of 86% up to 26 days after the dose.
Following oral administration of 14C-ivacaftor alone, the majority of ivacaftor (87.8%) was eliminated in the faeces after metabolic conversion.
For elexacaftor, tezacaftor and ivacaftor there was negligible urinary excretion of unchanged drug.
Hepatic impairment.
Elexacaftor alone or in combination with tezacaftor and ivacaftor has not been studied in subjects with severe hepatic impairment (Child-Pugh Class C, score 10-15). Following multiple doses of elexacaftor, tezacaftor and ivacaftor for 10 days, subjects with moderately impaired hepatic function (Child-Pugh Class B, score 7-9) had 25% higher AUC and 12% higher Cmax for elexacaftor, 73% higher AUC and 70% higher Cmax for M23-ELX, 36% higher AUC and 24% higher Cmax for combined ELX and M23-ELX, 20% higher AUC but similar Cmax for tezacaftor, and 50% higher AUC and 10% higher Cmax for ivacaftor compared with healthy subjects matched for demographics (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)).
Tezacaftor and ivacaftor.
Following multiple doses of tezacaftor and ivacaftor for 10 days, subjects with moderately impaired hepatic function had an approximately 36% higher AUC and 10% higher Cmax for tezacaftor, and a 1.5-fold higher AUC but similar Cmax for ivacaftor compared with healthy subjects matched for demographics.
Ivacaftor.
In a study with ivacaftor alone, subjects with moderately impaired hepatic function had similar ivacaftor Cmax, but an approximately 2.0-fold higher ivacaftor AUC0-∞ compared with healthy subjects matched for demographics.
Renal impairment.
Elexacaftor alone or in combination with tezacaftor and ivacaftor has not been studied in patients with severe renal impairment (eGFR less than 30 mL/min/1.73 m2) or in patients with end stage renal disease.
In human pharmacokinetic studies of elexacaftor, tezacaftor, and ivacaftor, there was minimal elimination of elexacaftor, tezacaftor, and ivacaftor in urine (only 0.23%, 13.7% [0.79% as unchanged drug], and 6.6% of total radioactivity, respectively).
Based on population pharmacokinetic (PK) analysis, exposure of elexacaftor was similar in patients with mild renal impairment (N=75, eGFR 60 to less than 90 mL/min/1.73 m2) relative to those with normal renal function (N=341, eGFR 90 mL/min/1.73 m2 or greater).
In population PK analysis conducted in 817 patients administered tezacaftor alone or in combination with ivacaftor in phase 2 or phase 3 studies indicated that mild renal impairment (N=172; eGFR 60 to less than 90 mL/min/1.73 m2) and moderate renal impairment (N=8; eGFR 30 to less than 60 mL/min/1.73 m2) did not affect the clearance of tezacaftor significantly (see Section 4.2 Dose and Method of Administration).
Special population.
Paediatric patients 2 to less than 18 years of age.
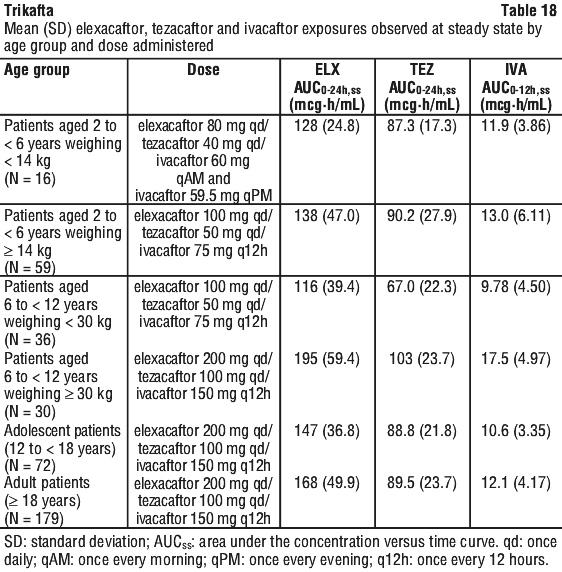
Elexacaftor, tezacaftor and ivacaftor exposures observed in phase 3 studies as determined using population PK analysis are presented by age group and dose administered in Table 18. Exposures of elexacaftor, tezacaftor and ivacaftor in patients 2 to less than 18 years of age are within the range observed in patients aged 18 years and older.
Gender.
Based on population PK analysis, the exposures of elexacaftor, tezacaftor and ivacaftor are similar in males and females.
5.3 Preclinical Safety Data
Genotoxicity.
Elexacaftor, tezacaftor and ivacaftor were all negative for genotoxicity in the following assays: Ames test for bacterial gene mutation, in vitro chromosomal aberration assay (in TK6 [human lymphoblastoid] cells for elexacaftor, and in Chinese hamster ovary cells for tezacaftor and ivacaftor), and in vivo bone marrow micronucleus test (performed in rats with elexacaftor, and in mice for tezacaftor and ivacaftor).
Carcinogenicity.
Elexacaftor was not carcinogenic in a 6-month study in transgenic (Tg.rasH2) mice, involving oral administration at doses up to 50 mg/kg/day (yielding systemic exposure 8-fold higher than in patients at the MRHD based on summed AUCs for elexacaftor and M23-ELX). No evidence of tumourigenicity was observed with elexacaftor in rats at oral doses up to 10 mg/kg/day for 92-93 weeks (yielding approximately 2 and 6 times the exposure in patients at the MRHD based on summed AUCs of elexacaftor and its M23 metabolite in male and female rats, respectively).
No evidence of tumourigenicity by tezacaftor was observed in a 6-month study in transgenic (Tg.rasH2) mice and in a conventional 2-year study in rats, conducted by the oral route. The highest doses tested (500 mg/kg/day in mice, 50 mg/kg/day in male rats and 75 mg/kg/day in female rats) yielded exposure to tezacaftor and its M1 and M2 metabolites that was 1.5-fold higher in mice, 1.2-fold higher in male rats, and 2.1-fold higher in female rats than in patients at the MRHD (based on summed AUCs).
Two-year oral studies in mice and rats demonstrated that ivacaftor was not carcinogenic in either species. Plasma exposures to ivacaftor in mice at the non-carcinogenic dosage (200 mg/kg/day, the highest dosage tested) were approximately 5- to 9-fold higher than the plasma levels measured in humans following Trikafta therapy, and at least 1.1- to 2.3-fold higher with respect to the summed AUCs for ivacaftor and its major metabolites. Plasma exposures to ivacaftor in rats at the non-carcinogenic dosage (50 mg/kg/day, the highest dosage tested) were approximately 20- to 36-fold higher than the plasma levels measured in humans following Trikafta therapy, and 6- to 9-fold higher with respect to the summed AUCs for ivacaftor and its major metabolites.6 Pharmaceutical Particulars
6.1 List of Excipients
Trikafta film-coated tablets.
Elexacaftor/tezacaftor/ivacaftor 100 mg/50 mg/75 mg or 50 mg/25 mg/37.5 mg).
Hypromellose, hypromellose acetate succinate, sodium lauryl sulfate, croscarmellose sodium, microcrystalline cellulose, magnesium stearate.
Elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg.
Opadry complete film coating system 20A130036 orange (ARTG PI No: 136412).
Elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37.5 mg.
Opadry complete film coating system 20A130039 orange (ARTG PI No: 141534).
Ivacaftor (150 mg or 75 mg).
Silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, carnauba wax.
Ivacaftor 150 mg.
Opadry II complete film coating system 85F90614 blue (ARTG PI No: 108371).
Opacode monogramming ink S-1-17823 black (ARTG PI No: 12108).
Ivacaftor 75 mg.
Opadry II complete film coating system 85F105098 blue (ARTG PI No: 140157).
Opacode monogramming ink S-1-17823 black (ARTG PI No: 12108).
Trikafta granules.
Elexacaftor/tezacaftor/ivacaftor (100 mg/50 mg/75 mg or 80 mg/40 mg/60 mg).
Silicon dioxide, croscarmellose sodium, hypromellose, hypromellose acetate succinate, lactose monohydrate, magnesium stearate, mannitol, sodium lauryl sulfate, sucralose.
Ivacaftor (75 mg or 59.5 mg).
Silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, lactose monohydrate, magnesium stearate, mannitol, sodium lauryl sulfate, sucralose.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf-life can be found on the public summary of the ARTG. The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
Store in original container.
6.5 Nature and Contents of Container
Tablets.
Thermoform blister consisting of PCTFE (polychlorotrifluoroethylene) film laminated to PVC (polyvinyl chloride) film and sealed with a blister foil lidding.
Pack sizes.
Trikafta [co-pack]: Pack size of 84 tablets (56 elexacaftor/tezacaftor/ivacaftor tablets and 28 ivacaftor tablets).
Granules.
Biaxially-oriented polyethylene terephthalate/polyethylene/foil/polyethylene (BOPET/PE/Foil/PE) printed foil laminate sachet.
Pack sizes.
Trikafta [co-pack]: Pack size of 56 sachets (4 weekly wallets, each with 7 elexacaftor/tezacaftor/ivacaftor sachets and 7 ivacaftor sachets).
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking it to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
 Elexacaftor: N-(1,3-dimethyl-1H-pyrazole-4-sulfonyl)-6-[3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide.
Elexacaftor: N-(1,3-dimethyl-1H-pyrazole-4-sulfonyl)-6-[3-(3,3,3-trifluoro-2,2-dimethylpropoxy)-1H-pyrazol-1-yl]-2-[(4S)-2,2,4-trimethylpyrrolidin-1-yl]pyridine-3-carboxamide.
 Tezacaftor: 1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-{1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1Hindol-5-yl}cyclopropane-1-carboxamide.
Tezacaftor: 1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-{1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1Hindol-5-yl}cyclopropane-1-carboxamide.
 Ivacaftor: N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo- 1,4-dihydroquinoline-3-carboxamide.
Ivacaftor: N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo- 1,4-dihydroquinoline-3-carboxamide.
CAS number.
Elexacaftor: 2216712-66-0.
Tezacaftor: 1152311-62-0.
Ivacaftor: 873054-44-5.7 Medicine Schedule (Poisons Standard)
Schedule 4.
Summary Table of Changes