What is in this leaflet?
Please read this leaflet carefully before you take TRIUMEQ. This leaflet answers some common questions about TRIUMEQ (dolutegravir/abacavir/lamivudine). It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the expected benefits of you taking TRIUMEQ against the risks this medicine could have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What is TRIUMEQ used for?
TRIUMEQ contains three active ingredients that are used to treat HIV infection in adults and children over 12 years old: dolutegravir, abacavir, and lamivudine. Dolutegravir belongs to a group of anti-retroviral medicines called integrase inhibitors (INIs). Abacavir and lamivudine belong to a group of anti-retroviral medicines called nucleoside analogue reverse transcriptase inhibitors (NRTIs).
TRIUMEQ does not cure HIV infection; it reduces the amount of virus in your body, and keeps it at a low level. TRIUMEQ also increases the CD4 cell count in your blood. CD4 cells are a type of white blood cells that are important in helping your body to fight infection.
To control your HIV infection, and to stop your illness from getting worse, you must keep taking all your medicines, unless your doctor tells you to stop taking any.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
TRIUMEQ is not addictive.
TRIUMEQ is not recommended for children under the age of 12 years as the dosage cannot be modified.
This medicine is available only with a doctor's prescription.
Before you take TRIUMEQ
When you must not take it
You must not take TRIUMEQ if:
- you have ever had an allergic reaction to abacavir, which is also included in medicines called KIVEXA, TRIZIVIR and ZIAGEN
- if you are allergic to the active ingredients dolutegravir or lamivudine, or any of the other ingredients listed toward the end of this leaflet.
- if you're taking another medicine called dofetilide, pilsicainide (to treat heart conditions) or fampridine (used in multiple sclerosis)
- if you have a serious liver disease TRIUMEQ may not be suitable for you.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering.
If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Special Warning - Hypersensitivity Reactions
TRIUMEQ contains abacavir and dolutegravir. Both of these active substances can cause a serious allergic reaction known as a hypersensitivity reaction.
Abacavir hypersensitivity reactions can be life-threatening if treatment with abacavir containing products is not stopped.
Research has found that people with a gene called HLA-B (type 5701) are more likely to have a hypersensitivity reaction to abacavir. However, even if you do not have this gene type it is still possible for you to get this reaction. If you know you have this gene type, be sure to tell your doctor before you take abacavir.
The most common symptoms of this reaction include high temperature (fever) and a skin rash. Other most frequently seen symptoms include nausea, vomiting, diarrhoea or abdominal pain; severe tiredness or body aches or generally feeling ill; headache; shortness of breath, sore throat or cough. If you develop any of these symptoms call your doctor IMMEDIATELY WHO WILL ADVISE YOU WHETHER YOU SHOULD STOP TAKING TRIUMEQ TABLETS. If your doctor is not available you must urgently seek other medical advice (e.g. the Accident and Emergency unit of the nearest hospital) before taking the next dose.
Other symptoms may include joint or muscle pain, swelling of the neck or itchy skin. Occasionally inflammation of the eye (conjunctivitis), ulcers in the mouth or low blood pressure may occur. The symptoms of this allergic reaction can occur at any time during treatment with TRIUMEQ tablets. However they usually occur in the first six weeks of treatment, and get worse with continued treatment.
If you have had this serious reaction to TRIUMEQ tablets, NEVER take TRIUMEQ or any other medicinal product containing abacavir (ZIAGEN, KIVEXA, TRIZIVIR) again as within hours you may experience a life-threatening lowering of your blood pressure or death.
Occasionally life threatening hypersensitivity reactions have occurred when medicine containing abacavir was restarted in patients who reported only one of the symptoms on the Alert Card before stopping.
On very rare occasions, hypersensitivity has been reported when treatment containing abacavir was re-started in patients who had no symptoms of hypersensitivity before stopping.
If you have stopped taking TRIUMEQ tablets for any reason it is important that you contact your doctor before restarting. This is especially so if you think you are having side-effects from other medicines or have another illness. Your doctor will check whether any symptoms you had before stopping may be related to this hypersensitivity reaction. If your doctor thinks there is a possibility that they were related, you may be told never to take TRIUMEQ tablets again. It is important that you follow this advice.
If you are hypersensitive to TRIUMEQ tablets you should return all of your unused TRIUMEQ tablets to your doctor or pharmacist for proper disposal.
The TRIUMEQ pack includes an Alert Card, to remind you and medical staff about abacavir hypersensitivity. Detach this card and keep it with you at all times.
Before you start to take it
Some other conditions may develop during HIV treatment.
Old infections may flare up
People with advanced HIV infection (AIDS) have weak immune systems, and are more likely to develop serious infections (opportunistic infections). When these people start treatment, they may find that old, hidden infections flare up, causing signs and symptoms of inflammation. These symptoms are probably caused by the body's immune system becoming stronger, so that the body starts to fight these infections.
If you get any symptoms of infection while you're taking TRIUMEQ:
Tell your doctor immediately. Don't take other medicines for the infection without your doctor's advice.
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
If you are pregnant, or think you could be, or if you are planning to have a baby, don't take TRIUMEQ without checking with your doctor.
Your doctor will consider the benefit to you and the risk to your baby of taking TRIUMEQ while you're pregnant
If you could get pregnant while taking TRIUMEQ, you need to use a reliable method of contraception to prevent pregnancy. Taking dolutegravir (one of the components of TRIUMEQ) at the time of becoming pregnant, or during the first twelve weeks of pregnancy, may increase the risk of a type of birth defect, called neural tube defect, such as spina bifida (malformed spinal cord).
Your doctor can discuss with you the risks and benefits involved.
Abacavir and lamivudine belong to a group of anti-retroviral medicines called nucleoside analogue reverse transcriptase inhibitors (NRTIs). In babies and infants exposed to NRTIs during pregnancy or labour small temporary increases in blood levels of a substance called lactate have been observed. Additionally there have been very rare reports of diseases that affect the nervous system such as a delayed development and seizures. Overall, in children whose mothers took NRTIs during pregnancy, the benefit from the reduced chance of being infected with HIV is likely to be greater than the risk of suffering from side effects.
Where possible, women who are HIV-positive should not breast feed. This is because HIV infection can be passed on to the baby in breast milk. If formula feeding is not possible, you should get advice from your doctor.
A small amount of the ingredients in TRIUMEQ can also pass into your breast-milk.
If you have not told your doctor about any of the above, tell him/her before you start taking TRIUMEQ.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Don't take TRIUMEQ with these medicines:
- dofetilide or pilsicainide, to treat heart conditions
- fampridine, used in multiple sclerosis
These medicines should not be used with TRIUMEQ:
- emtricitabine, to treat HIV infection
- sorbitol-containing medicines (usually liquids) used regularly
Tell your doctor if you're being treated with any of these.
Methadone and TRIUMEQ
If you are taking methadone, your doctor may need to adjust your methadone dose, as abacavir (one of the active substances in TRIUMEQ) increases the rate at which methadone leaves your body. This is unlikely to affect most methadone users.
Tell your doctor if you are taking:
- metformin, to treat diabetes
- medicines called antacids, to treat indigestion and heartburn. Do not take an antacid that contains aluminium, calcium or magnesium during the 6 hours before you take TRIUMEQ, or for at least 2 hours after you take it. These antacids, readily available over the counter, will decrease the effectiveness of TRIUMEQ if taken at the same time as TRIUMEQ.
- calcium and iron supplements. Do not take a calcium or iron supplement during the 6 hours before you take TRIUMEQ, or for at least 2 hours after you take it. If you take food with your medicine you can take a calcium or iron supplement at the same time as TRIUMEQ.
- etravirine, efavirenz, fosamprenavir/ritonavir, nevirapine or tipranavir/ritonavir, to treat HIV infection
- rifampicin, to treat tuberculosis (TB) and other bacterial infections
- co-trimoxazole, an antibiotic used to treat Pneumocystis jiroveci pneumonia (often referred to as PCP) or toxoplasmosis
- phenytoin and phenobarbital, to treat epilepsy
- carbamazepine, to treat epilepsy and bipolar disorder
- St. John's wort, (Hypericum perforatum), a herbal remedy to treat depression
Tell your doctor or pharmacist if you are taking any of these. Your doctor may decide to adjust your dose or that you need extra checkups.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How do I take TRIUMEQ?
Follow all directions given to you by your doctor or pharmacist carefully, and take great care not to miss any doses if at all possible.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
The usual dose of TRIUMEQ in adults and in children over the age of 12 years weighing at least 40 kg is one combined tablet (50 mg dolutegravir, 600 mg abacavir, 300 mg lamivudine) taken once a day.
If you weigh less than 40kg, you cannot take TRIUMEQ, because the dose of each component of this medicine cannot be adjusted to your weight. Your doctor might prescribe the components separately for you.
How to take it
Swallow the tablets whole with a full glass of water.
Antacid medicines
Antacids which contain calcium, aluminium or magnesium will reduce the effectiveness of TRIUMEQ during the 6 hours before you take TRIUMEQ, or for at least 2 hours after you take it.
Other acid-lowering medicines like ranitidine and omeprazole can be taken at the same time as TRIUMEQ. TALK TO YOUR DOCTOR FOR FURTHER ADVICE BEFORE TAKING ACID-LOWERING MEDICINES WITH TRIUMEQ.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it for
Continue taking your medicine for as long as your doctor tells you.
If you forget to take it
If you miss a dose, take it as soon as you remember. But if your next dose is due within 4 hours, skip the dose you missed and take the next one at the usual time. Then continue your treatment as before.
Don't take a double dose to make up for a missed dose. Just take it as soon as you remember.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (In Australia call 131126. In New Zealand call 0800 POISON or 0800 764 766) for advice if you think you or anyone else may have taken too much TRIUMEQ, even if there are no signs of discomfort or poisoning. If you are not sure what to do, contact your doctor or pharmacist. You may need urgent medical attention.
Don't stop TRIUMEQ without advice
Take TRIUMEQ for as long as your doctor recommends. Don't stop unless your doctor advises you to.
If you have stopped taking TRIUMEQ for any reason, particularly because you think you are having side effects or for other illness, it is important that you contact your doctor before restarting. In some cases your doctor will ask you to restart TRIUMEQ in a place where you will be able to get ready access to medical care if needed.
If you have hepatitis B infection, you should not stop TRIUMEQ tablets without instructions from your doctor, as you may have a recurrence of your hepatitis. This may occur due to you suddenly stopping lamivudine.
While you are using TRIUMEQ
You will need regular blood tests
For as long as you're taking TRIUMEQ, your doctor will arrange regular blood tests to check for side effects.
Stay in regular contact with your doctor
TRIUMEQ helps to control your condition, but it is not a cure for HIV infection. You need to keep taking it every day to stop your illness from getting worse. Because TRIUMEQ does not cure HIV infection, you may still develop other infections and illnesses linked to HIV infection.
Keep in touch with your doctor, and don't stop taking TRIUMEQ without your doctor's advice.
Protect other people
HIV infection is spread by sexual contact with someone who has the infection, or by transfer of infected blood (for example, by sharing injection needles). You can still pass on HIV when taking this medicine, although the risk is lowered by effective antiretroviral therapy. Discuss with your doctor the precautions needed to avoid infecting other people, which may include:
- Use a condom when you have oral or penetrative sex.
- Don't risk blood transfer - for example, don't share needles.
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking TRIUMEQ.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
TRIUMEQ can make you dizzy and have other side effects that make you less alert.
Don't drive or use machines unless you are sure you're not affected.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TRIUMEQ.
This medicine helps most people with HIV, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
When you're being treated for HIV, it can be hard to tell whether a symptom is a side effect of TRIUMEQ or other medicines you are taking, or an effect of the HIV disease itself. So it is very important to talk to your doctor about any changes in your health.
Some side effects may only be seen in your blood tests and may not appear immediately after you start taking TRIUMEQ. If you get any of these effects, and if they are severe, your doctor may advise you to stop taking TRIUMEQ.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Hypersensitivity reactions
TRIUMEQ contains abacavir and dolutegravir. Both of these active substances can cause a serious allergic reaction known as a hypersensitivity reaction.
These hypersensitivity reactions have been seen more frequently in people taking medicines that contain abacavir.
Who gets these reactions?
Anyone taking TRIUMEQ could develop a hypersensitivity reaction, which could be life threatening if they continue to take TRIUMEQ.
You are more likely to develop such a reaction if you have a gene called HLA-B*5701 (but you can get a reaction even if you don't have this gene). If possible, you will have been tested for this gene before TRIUMEQ was prescribed for you. If you know you have this gene, tell your doctor before you take TRIUMEQ.
What are the symptoms?
The most common symptoms are:
- fever (high temperature) and skin rash.
Other common symptoms are:
- nausea (feeling sick), vomiting (being sick), diarrhoea, abdominal (stomach) pain, severe tiredness, shortness of breath, cough, headache, muscle pain and discomfort.
Other less common symptoms can include:
- pains in the joints, swelling of the neck, serious breathing problems, sore throat
- occasionally, inflammation of the eye (conjunctivitis), mouth ulcers, low blood pressure, tingling or numbness of the hands or feet.
When do these reactions happen?
Hypersensitivity reactions can start at any time during treatment with TRIUMEQ but are more likely during the first 6 weeks of treatment.
Contact your doctor immediately:
- if you get a skin rash, OR
- if you get symptoms from at least 2 of the following groups:
- fever
- shortness of breath, sore throat or cough
- nausea or vomiting, diarrhoea or stomach pain
- severe tiredness or achiness, or generally feeling ill.
Your doctor may advise you to stop taking TRIUMEQ.
Always carry your Alert Card while you are taking TRIUMEQ.
If you have stopped taking TRIUMEQ:
If you have stopped taking TRIUMEQ because of a hypersensitivity reaction, you must NEVER AGAIN take TRIUMEQ, or any other medicine containing abacavir (ZIAGEN, KIVEXA or TRIZIVIR). If you do, within hours, your blood pressure could fall dangerously low, which could result in death. You should also never again take medicines containing dolutegravir.
If you have stopped taking TRIUMEQ for any reason, especially because you think you are having side effects, or because you have another illness:
Talk to your doctor before you start again. Your doctor will check whether your symptoms were related to a hypersensitivity reaction. If the doctor thinks they may have been, you will then be told never again to take TRIUMEQ, or any other medicine containing abacavir. You may also be told never again to take any other medicine containing dolutegravir. It is important that you follow this advice.
Occasionally, reactions have developed in people who start taking abacavir again and had only one symptom on the Alert Card before they stopped taking it.
Very rarely, reactions have developed in people who start taking abacavir again, but who had no symptoms before they stopped taking it.
If your doctor advises that you can start taking TRIUMEQ again, you may be asked to take your first doses in a place where you will have ready access to medical care if you need it.
If you are hypersensitive to TRIUMEQ, return all your unused TRIUMEQ tablets for safe disposal. Ask your doctor or pharmacist for advice.
The TRIUMEQ pack includes an Alert Card, to remind you and medical staff about hypersensitivity reactions. Detach this card and keep it with you at all times.
Symptoms of infection and inflammation
People with advanced HIV infection (AIDS) have weak immune systems and are more likely to develop serious infections (opportunistic infections). When they start treatment, the immune system becomes stronger, so the body starts to fight infections.
Symptoms of infection and inflammation may develop, caused by either:
- old, hidden infections flaring up again as the body fights them
- the immune system attacking healthy body tissue (autoimmune disorders)
The symptoms of autoimmune disorders may develop many months after you start taking medicine to treat your HIV infection.
Symptoms may include:
- muscle weakness and/or muscle pain
- joint pain or swelling
- weakness beginning in the hands and feet and moving up towards the trunk of the body
- palpitations or tremor
- hyperactivity (excessive restlessness and movement).
If you get any symptoms of infection while you're taking TRIUMEQ:
Tell your doctor immediately. Don't take other medicines for the infection without your doctor's advice.
Fat loss or fat gain
Fat loss or fat gain has been observed with combined antiretroviral therapy. A causal relationship for this has not been established. Should any change in body shape be noticed, seek medical advice.
Lactic acidosis is a rare but serious side effect
Some people taking TRIUMEQ, or other medicines like it (NRTIs), develop a condition called lactic acidosis, together with an enlarged liver.
Lactic acidosis is caused by a build-up of lactic acid in the body. It is rare; if it happens, it usually develops after a few months of treatment. It can be life-threatening, causing failure of internal organs.
Lactic acidosis is more likely to develop in people who have liver disease, especially in women.
Signs of lactic acidosis include:
- deep, rapid, difficult breathing
- drowsiness
- numbness or weakness in the limbs
- feeling sick (nausea), being sick (vomiting)
- stomach pain.
During your treatment, your doctor will monitor you for signs of lactic acidosis. If you have any of the symptoms listed above or any other symptoms that worry you see your doctor as soon as possible.
As well as the conditions listed above, other side effects can develop during combination therapy for HIV.
Very common side effects
These may affect more than 1 in 10 people:
- headache
- diarrhoea
- feeling sick (nausea)
Common side effects
These may affect up to 1 in 10 people:
- being sick (vomiting)
- stomach pains (abdominal pain) and bloating (abdominal distension)
- stomach (abdominal) discomfort
- wind (flatulence)
- indigestion (dyspepsia)
- gastro-oesophageal reflux disease
- loss of appetite
- feeling drowsy
- tiredness, lack of energy
- general feeling of being unwell
- high temperature (fever)
- dizziness
- nightmares and abnormal dreams
- sleep disorder
- depression (feelings of deep sadness and unworthiness)
- anxiety
- hair loss
- abacavir hypersensitivity reaction (See hypersensitivity reactions' earlier in this section)
- skin rash
- itching (pruritis)
- joint pain
- muscle pain and discomfort
Common side effects that may show up in blood tests are:
- increase in triglycerides (type of fat) in the blood
- increase in glucose (sugar) in the blood
Uncommon side effects
These may affect up to 1 in 100 people:
- dolutegravir allergic reaction (See 'allergic reactions' earlier in this section)
- inflammation of the liver (hepatitis)
- suicidal thoughts and behaviours (mainly in patients who have had depression or mental health problems before)
- weight gain
Uncommon side effects that may show up in blood tests are:
- a low red blood cell count (anaemia) or low white blood cell count (neutropenia)
- a decrease in the number of cells involved in blood clotting (thrombocytopenia)
- an increase in the level of liver enzymes
Rare side effects
These may affect up to 1 in 1000 people:
- lactic acidosis (see 'lactic acidosis is a rare but serious side effect' earlier in this section)
- liver failure (signs may include yellowing of the skin and the whites of the eyes or unusually dark urine)
- inflammation of the pancreas (pancreatitis)
- breakdown of muscle tissue
Rare side effects that may show up in blood tests are:
- increase in an enzyme called amylase
Very rare side effects
These may affect up to 1 in 10,000 people:
- tingling or numbness of the hands and feet (paraesthesiae)
- numbness, tingling or weakness of the arms and legs (peripheral neuropathy)
- skin rash, which may form blisters and looks like small targets (central dark spots surrounded by a paler area, with a dark ring around the edge) (erythema multiforme)
- widespread rash with blisters and peeling skin, particularly around the mouth, nose, eyes and genitals (Stevens-Johnson syndrome), and a more severe form causing skin peeling in more than 30% of the body surface (toxic epidermal necrolysis).
Very rare side effects that may show up in blood tests are:
- failure of the bone marrow to produce new red blood cells (pure red cell aplasia)
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using TRIUMEQ
Storage
Store in the original package in order to protect from moisture. Keep the bottle tightly closed. Do not remove the desiccant.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store TRIUMEQ or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
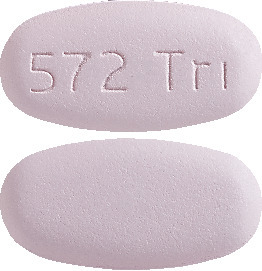
TRIUMEQ tablets are purple, oval, biconvex film-coated tablets debossed with '572 Trı' on one side and are available in bottles of 30 tablets with child-resistant closure.
Ingredients
TRIUMEQ contains 50 mg of dolutegravir (as dolutegravir sodium), 600 mg of abacavir (as abacavir sulfate) and 300 mg lamivudine as the active ingredients.
TRIUMEQ tablets also contain the following inactive ingredients:
- mannitol
- microcrystalline cellulose
- povidone
- sodium starch glycolate
- magnesium stearate
- polyvinyl alcohol
- titanium dioxide
- macrogol 3350
- talc
- iron oxide red
- iron oxide black
Supplier
ViiV Healthcare Pty Ltd
Level 4, 436 Johnston St,
Abbotsford, Victoria, 3067
Australia.
Where to go for further information:
Pharmaceutical companies are not in a position to give people an individual diagnosis or medical advice. Your doctor or pharmacist is the best person to give you advice on the treatment of your condition. You may also be able to find general information about your disease and its treatment from patient information groups and product specific organisations.
Trade marks are owned by or licensed to the ViiV Healthcare group of companies.
This leaflet was prepared on 29 October 2019
TRIUMEQ tablets: AUST R 218644
© 2019 ViiV Healthcare group of companies or its licensor.
Version 11.0
Published by MIMS August 2021




 The adverse reactions observed in the subset of patients who received dolutegravir + ABC/3TC in SPRING-2 and FLAMINGO were generally consistent with those seen for the overall patient population participating in these trials.
The adverse reactions observed in the subset of patients who received dolutegravir + ABC/3TC in SPRING-2 and FLAMINGO were generally consistent with those seen for the overall patient population participating in these trials.
 The rates of laboratory abnormalities and mean change in fasted lipid values remained generally similar between the 48 and 96 week data evaluation in SINGLE.
The rates of laboratory abnormalities and mean change in fasted lipid values remained generally similar between the 48 and 96 week data evaluation in SINGLE.
 In the primary 48 weeks analysis in the SINGLE study, the proportion of patients with virologic suppression (HIV-1 RNA < 50 copies/mL using a missing or discontinuation = failure analysis) in the dolutegravir + ABC/3TC arm (88%), was superior to the EFV/TDF/FTC arm (81%), p = 0.003. Similar treatment difference was observed in patients defined by baseline HIV-RNA level (< or > 100,000 copies/mL). The median time to viral suppression was 28 days in the group receiving dolutegravir + ABC/3TC and 84 days in the EFV/TDF/FTC arm (p < 0.0001). The adjusted mean change in CD4+ T cell count from baseline were 267 cells/mm3 in the group receiving dolutegravir + ABC/3TC and 208 cells/mm3 for the EFV/TDF/FTC arm in SINGLE at 48 week [adjusted difference between arm (with 95% CI), 58.9 cells (33.4 cells to 84.4 cells), p < 0.001]. Both the time to viral suppression and change from baseline analyses were pre-specified and adjusted for multiplicity.
In the primary 48 weeks analysis in the SINGLE study, the proportion of patients with virologic suppression (HIV-1 RNA < 50 copies/mL using a missing or discontinuation = failure analysis) in the dolutegravir + ABC/3TC arm (88%), was superior to the EFV/TDF/FTC arm (81%), p = 0.003. Similar treatment difference was observed in patients defined by baseline HIV-RNA level (< or > 100,000 copies/mL). The median time to viral suppression was 28 days in the group receiving dolutegravir + ABC/3TC and 84 days in the EFV/TDF/FTC arm (p < 0.0001). The adjusted mean change in CD4+ T cell count from baseline were 267 cells/mm3 in the group receiving dolutegravir + ABC/3TC and 208 cells/mm3 for the EFV/TDF/FTC arm in SINGLE at 48 week [adjusted difference between arm (with 95% CI), 58.9 cells (33.4 cells to 84.4 cells), p < 0.001]. Both the time to viral suppression and change from baseline analyses were pre-specified and adjusted for multiplicity. Limited data are available in adolescents receiving a daily dose of 600 mg of abacavir and 300 mg of lamivudine. Pharmacokinetic parameters are comparable to those reported in adults.
Limited data are available in adolescents receiving a daily dose of 600 mg of abacavir and 300 mg of lamivudine. Pharmacokinetic parameters are comparable to those reported in adults.


