What is in this leaflet
This leaflet answers some common questions about Truxima® intravenous infusion. It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given Truxima® against the benefits they expect it will have for you.
If you have any concerns about being given this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
What Truxima® is used for
Truxima® contains the active ingredient rituximab, which is a monoclonal antibody. Monoclonal antibodies are proteins which specifically recognise and bind to other unique proteins in the body.
Truxima® is used to treat rheumatoid arthritis (RA), Granulomatosis with polyangiitis (Wegener's) (GPA) and Microscopic polyangiitis (MPA).
RA is an inflammatory disease of the joints. GPA and MPA are inflammatory diseases of the blood vessels.
Truxima® works by binding to a protein on the surface of certain white blood cells known as B lymphocytes. B lymphocytes play a role in the inflammation observed in RA, GPA and MPA. By binding to the protein Truxima® reduces the ability of B lymphocytes to cause inflammation.
In RA Truxima® can also slow down the damage to your joints and improve your ability to do your normal daily activities.
Your doctor may have prescribed Truxima® for another reason.
Ask your doctor if you have any questions why Truxima® has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you are given Truxima®
When you must not be given Truxima®
Do not use Truxima®:
- if you have had an allergic reaction to rituximab or any of the ingredients listed at the end of this leaflet
- if you have had an allergic reaction to any other proteins that are of mouse origin
Some of the symptoms of an allergic reaction may include severe skin rash, itching, hives, swelling of the face, lips, mouth or throat which may cause difficulty in swallowing or breathing, swelling of the hands, feet or ankles.
If you are not sure if you should start receiving Truxima®, talk to your doctor.
Before you are given Truxima®
Your doctor must know about all the following before you are given Truxima®.
Tell your doctor if:
- you have an infection, or a history of a recurring or long- term infection such as hepatitis B
- you are taking or have previously taken medicines which may affect your immune system, such as chemotherapy or immunosuppressive medicines
If you are taking or have taken medicines which affect your immune system, you may have an increased risk of infections. There have been reports of a rare, serious brain infection called PML (progressive multifocal leukoencephalopathy) usually affecting people with a weakened immune system. Your chance of getting PML may be higher if you are treated with Truxima® and/or other medicines that weaken the immune system. PML can cause severe disability or even death.
- you have a history of heart disease with:
- angina
- cardiac arrhythmias (abnormal beating of the heart)
- congestive heart failure
Your doctor will supervise you closely during treatment with Truxima®.
- you are taking medicine to control blood pressure
Truxima® may cause a reduction in blood pressure at the beginning of treatment. Because Truxima® may cause a temporary drop in your blood pressure, your doctor may advise you to temporarily stop taking your blood pressure medicine before you are given Truxima®.
- you have pre-existing lung disease
You may have a greater chance of breathing difficulties during treatment with Truxima®.
- you are 65 years of age or older or suffer from kidney problems and are receiving Truxima® for GPA or MPA
You may have an increased risk of serious side effects. Your doctor will monitor you closely during your treatment.
- you intend to have or have had immunisation with any vaccine (e.g. measles, rubella, flu, vaccines for travel purposes)
Some vaccines should not be given at the same time as Truxima® or in the months after you receive Truxima®. Your doctor will check if you should have any vaccines before you receive Truxima®.
- you are allergic to any other medicines or any other substances such as foods, preservatives or dyes
- you are pregnant or intend to become pregnant
It is not known whether Truxima® is harmful to an unborn baby. It is not recommended that you are given Truxima® while you are pregnant.
If you are a woman of child bearing potential, you must use effective contraceptive methods to prevent pregnancy during treatment and for 12 months after completing treatment with Truxima®.
- you are breast feeding or plan to breast feed.
It is not known if Truxima® passes into breast milk. It is recommended that you discontinue breast feeding while you are treated with Truxima®.
If you have not told your doctor about any of the above, tell them before you are given Truxima®.
Use in children
The safety and effectiveness of Truxima® have not been established in children.
Taking other medicines
Tell your doctor if you are taking any other medicines including any that you have bought without a prescription from a pharmacy, supermarket or healthfood shop.
As Truxima® may cause a temporary drop in your blood pressure at the beginning of treatment, your doctor may advise you to temporarily stop taking your blood pressure medicine before you are given Truxima®.
It is not known if Truxima® will affect your normal response to a vaccine
It is possible that after treatment with Truxima® you may experience allergic reactions if you are treated with other medications containing monoclonal antibodies.
Your doctor and pharmacist will have more information on medicines to be careful with or to avoid while undergoing treatment with Truxima®.
How Truxima® is given
Truxima® is given by slow infusion into a vein (intravenous infusion) by a healthcare professional. Before the infusion is given you will receive medicines to reduce the chance of any reactions to Truxima®.
The dose of Truxima® for treatment of RA is 1000 mg followed by a second dose of 1000 mg 2 weeks later. For RA Truxima® should be used together with methotrexate.
Depending on the circumstances of your disease or response to the drug, your doctor may decide to re-treat your RA with an additional course of Truxima®.
The dose of Truxima® for GPA and MPA patients is 375 mg per square metre of body surface area once a week for 4 weeks. For GPA or MPA Truxima® should be used together with glucocorticoids.
Overdose
As Truxima® is given to you under the supervision of your doctor, it is very unlikely that you will receive too much. However, if you experience any side effects after being given Truxima®, tell your doctor immediately.
While you are receiving Truxima®
Things you must do
If you are a woman of child bearing potential, you must use effective contraceptive methods to prevent pregnancy during treatment with Truxima® and for 12 months after completing treatment.
Tell your doctor if you become pregnant while receiving Truxima®.
Tell all doctors, dentists and pharmacists who are treating you that you are receiving Truxima®.
Tell your partner or caregiver you are receiving Truxima® and ask them to tell you if they notice any changes in your movement or behaviour. If they notice any changes you should tell your doctor about them immediately. Your doctor may need to perform some tests and alter your treatment.
Be sure to keep all your appointments with your doctor so that your progress can be checked. Your doctor will perform regular blood tests.
Things you must not do
Do not breast feed your infant during treatment with Truxima®. It is not known whether Truxima® crosses into human milk.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting with a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how Truxima® affects you. Truxima® generally does not cause any problems with your ability to drive or operate machinery. However, as with many other medicines, Truxima® may cause dizziness in some people.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are receiving Truxima®.
Do not be alarmed by this list of possible side effects. You may not experience any of them. Truxima® helps many people with RA, GPA or MPA but it may have unwanted side effects.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
The following is a list of the more common side effects.
During or after an infusion
Tell your doctor if you notice any of the following during or after receiving an infusion (particularly during the first 2 hours of receiving the first infusion) and they worry you:
- fever, chills and severe shivering (most likely to occur)
- swelling of the tongue, face, lips, mouth or throat which may cause difficulty breathing or swallowing
- itchy rash and/or pinkish, itchy swellings on the skin
- difficulty breathing and/or shortness of breath
- wheezing or coughing
- dizziness or lightheadedness, especially on standing up
- high blood pressure
- tremor
- nausea (feeling sick) or vomiting
- headache
- fatigue (feeling tired) and/or feeling weak
- rhinitis (a runny nose)
- flushing
- fast heart beat
- chest pain
- stomach pain or discomfort
- throat irritation
These side effects are temporary and less likely to occur after the first infusion.
Your doctor may recommend that you take medication to prevent pain or allergy before you receive your Truxima® infusion.
The following is a list of other side effects which may occur during treatment with Truxima®. Tell your doctor if you notice any of them and they worry you:
- aching muscles, muscle tenderness or weakness, muscle spasms
- indigestion
- painful, swollen joints
- severe headache
- high cholesterol
- insomnia
- diarrhoea
- pins and needles, or decreased feeling in the skin
- infections e.g. urinary tract infections, colds or chest infections including pneumonia
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- infections with fever, severe chills, sore throat or mouth ulcers
- severe skin rash, itching, hives
- swelling of the face, lips, mouth or throat which may cause difficulty in swallowing or breathing, swelling of the hands, feet or ankles
- one or a combination of the following: severe shortness of breath, severe difficulty breathing, severe wheezing, severe coughing
- severe stomach pain, nausea or vomiting
- vision loss associated with headaches, confusion and seizures
- one or a combination of the following: confusion, disorientation or memory loss, changes in the way you move, walk or talk, decreased strength or progressive weakness in your body, blurred or loss of vision
- yellowing of skin and eyes, light coloured bowel motions, dark coloured urine
- fever, headache and stiff neck, incoordination (ataxia), personality change, hallucinations, altered consciousness, seizures or coma – these may be due to a serious brain infection (enteroviral meningoencephalitis), which can be fatal.
These are serious side effects. You may need urgent medical attention. Serious side effects are rare.
This is not a complete list of all possible side effects. Your doctor or pharmacist has a more complete list. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell.
Ask your doctor or pharmacist if you don’t understand anything in this list.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After receiving Truxima®
Storage
Truxima® should be stored in the pharmacy or on the hospital ward.
The concentrated solution for infusion should be kept in a refrigerator at 2-8°C. It should not be frozen.
Truxima® should be stored away from light.
Product Description
Availability
Truxima® is available as 100 mg/10 mL and 500 mg/50 mL single dose vials.
Truxima® comes in packs of two vials for the 100mg/10 mL presentation and packs of one vial for the 500 mg/50 mL presentation.
What Truxima® looks like
Truxima® is available as a clear, colourless, concentrated solution for intravenous infusion. It is diluted before infusion into a vein.
Ingredients
Truxima® contains the active ingredient rituximab (rch). Truxima® comes in two strength, 100 mg and 500 mg. Each vial of Truxima® also contains the following inactive ingredients:
- sodium citrate
- polysorbate 80
- sodium chloride
- sodium hydroxide and/or hydrochloric acid
Sponsor
Celltrion Healthcare Australia Pty Ltd
Suite 13.03, 31 Market Street,
Sydney 2000, Australia
1800 325 228
Please check with your pharmacist for the latest Consumer Medicine Information.
Australian Registration Numbers:
- 100 mg vial: AUST R 285816
- 500 mg vial: AUST R 285815
This leaflet was prepared on October 2023
Published by MIMS February 2024

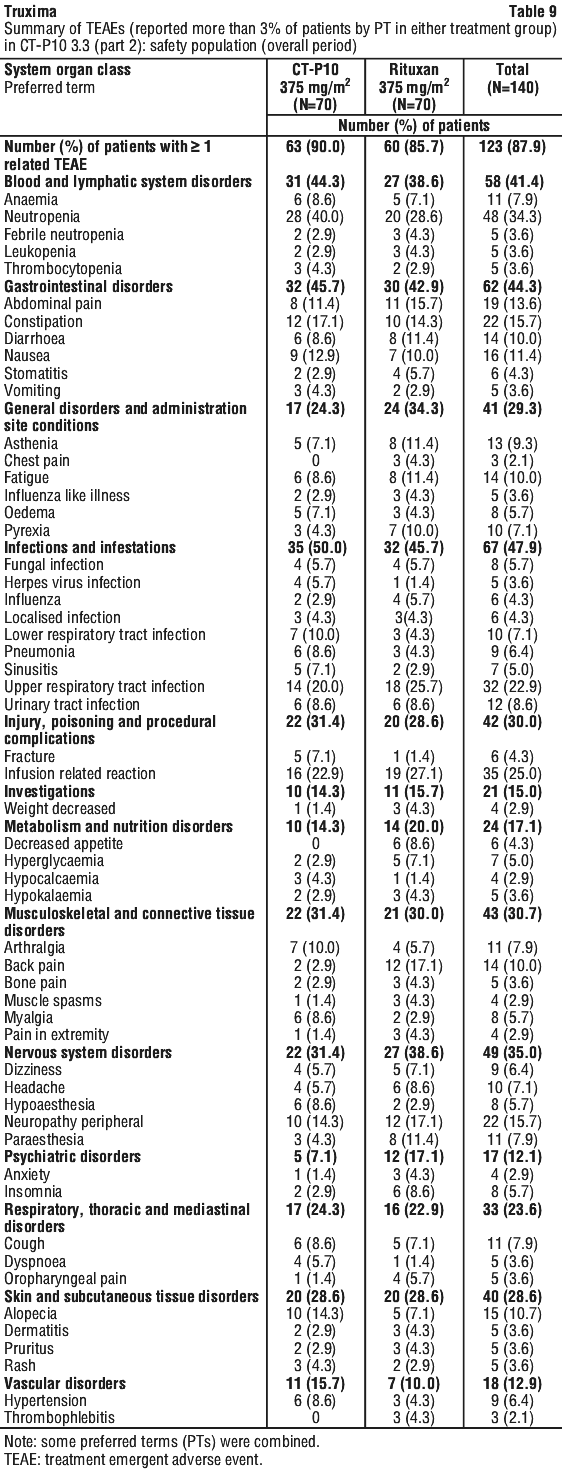
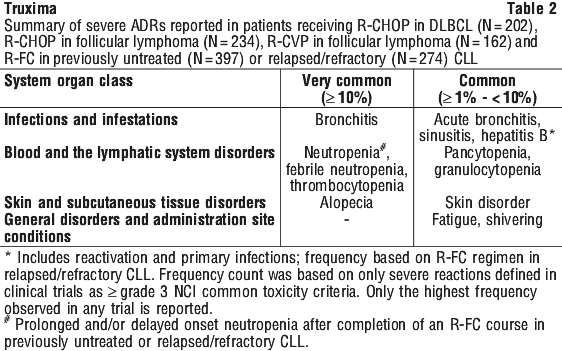 The following terms have been reported as adverse events, however, were reported at a similar (< 2% difference between the groups) or lower incidence in the rituximab-arms compared to control arms: haematotoxicity, neutropenic infection, urinary tract infection, septic shock, superinfection lung, implant infection, septicaemia staphylococcal, lung infection, rhinorrhoea, pulmonary oedema, cardiac failure, sensory disturbance, venous thrombosis, mucosal inflammation nos, influenza-like illness, oedema lower limb, abnormal ejection fraction, pyrexia, general physical health deterioration, fall, multi-organ failure, venous thrombosis deep limb, positive blood culture, diabetes mellitus inadequate control.
The following terms have been reported as adverse events, however, were reported at a similar (< 2% difference between the groups) or lower incidence in the rituximab-arms compared to control arms: haematotoxicity, neutropenic infection, urinary tract infection, septic shock, superinfection lung, implant infection, septicaemia staphylococcal, lung infection, rhinorrhoea, pulmonary oedema, cardiac failure, sensory disturbance, venous thrombosis, mucosal inflammation nos, influenza-like illness, oedema lower limb, abnormal ejection fraction, pyrexia, general physical health deterioration, fall, multi-organ failure, venous thrombosis deep limb, positive blood culture, diabetes mellitus inadequate control.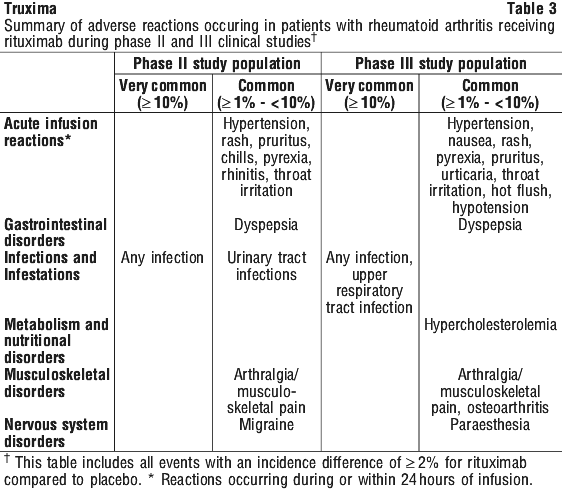 The following adverse events were reported at a frequency between 1% and 2% greater in the rituximab-arms compared to control arms: lower respiratory tract infections/pneumonia, abdominal pain upper, muscle spasms, asthenia.
The following adverse events were reported at a frequency between 1% and 2% greater in the rituximab-arms compared to control arms: lower respiratory tract infections/pneumonia, abdominal pain upper, muscle spasms, asthenia.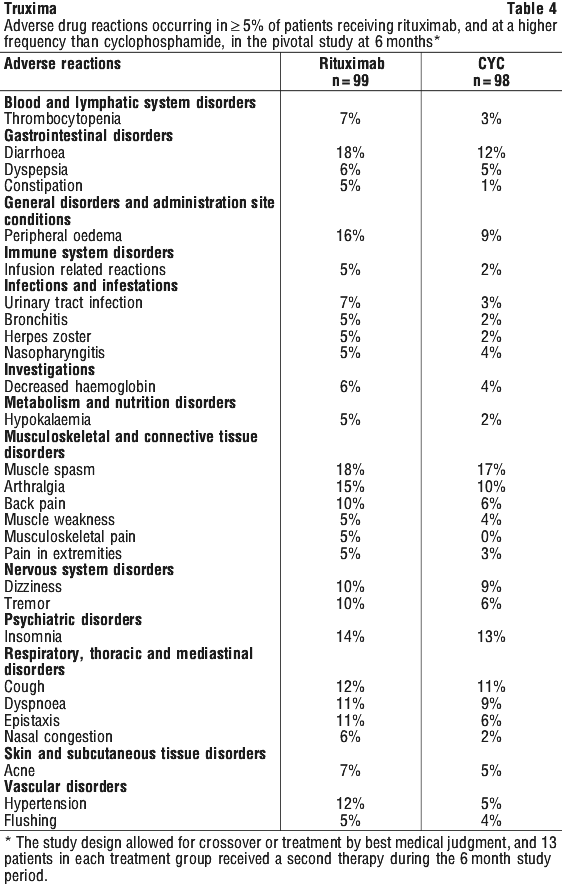 There was a higher incidence and rates of severe (Grade ≥ 3) and serious adverse events in older patients (aged ≥ 65 years) compared to younger patients (aged < 65 years), primarily attributable to anaemia and leucopenia, gastrointestinal disorders, and administrational site reactions. Deaths only occurred in older patients, with a similar incidence in the two treatment groups. Hospitalisations related to disease or study drug (per investigator's opinion) occurred more frequently in older patients in the rituximab group, with no hospitalisations occurring in older patients in the CYC group. There was no clear or consistent trend in the rates of infections and serious infections in younger patients versus older patients in either treatment group. (See Section 4.4 Special Warnings and Precautions for Use).
There was a higher incidence and rates of severe (Grade ≥ 3) and serious adverse events in older patients (aged ≥ 65 years) compared to younger patients (aged < 65 years), primarily attributable to anaemia and leucopenia, gastrointestinal disorders, and administrational site reactions. Deaths only occurred in older patients, with a similar incidence in the two treatment groups. Hospitalisations related to disease or study drug (per investigator's opinion) occurred more frequently in older patients in the rituximab group, with no hospitalisations occurring in older patients in the CYC group. There was no clear or consistent trend in the rates of infections and serious infections in younger patients versus older patients in either treatment group. (See Section 4.4 Special Warnings and Precautions for Use).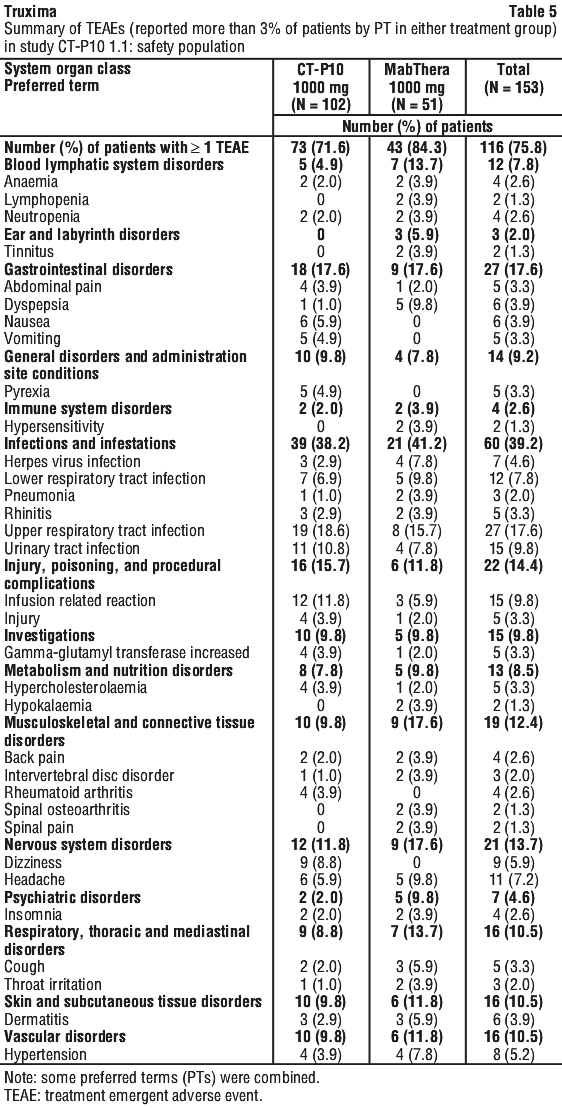
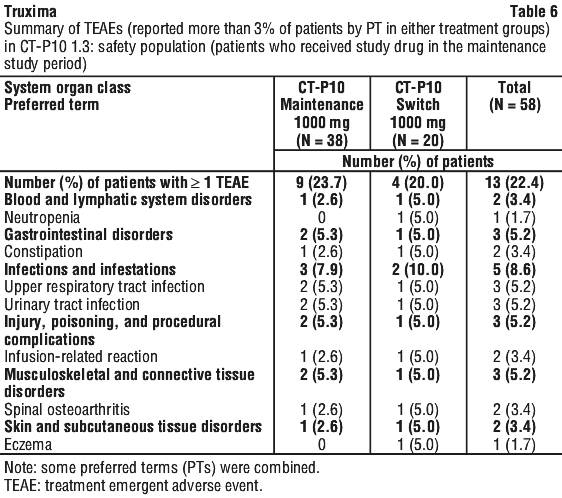
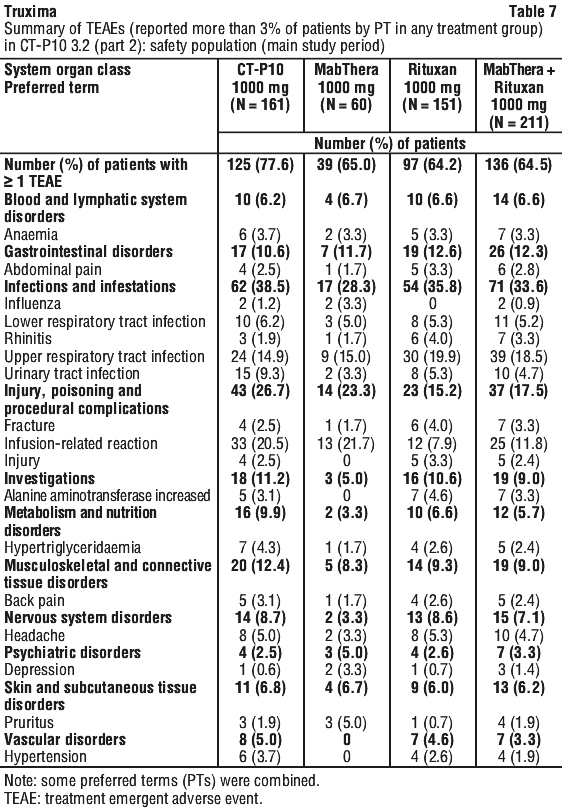
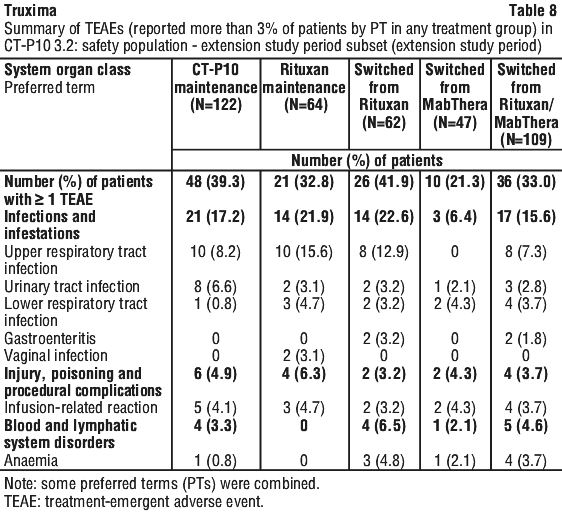
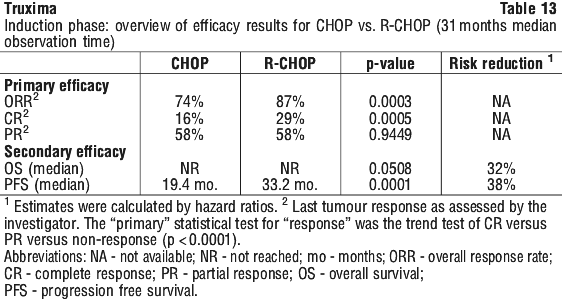
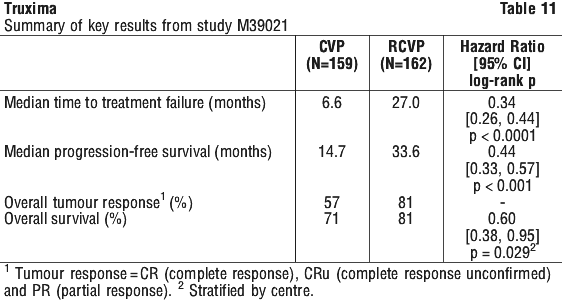 Results from three other randomised studies using rituximab in combination with chemotherapy regimens other than CVP (CHOP, MCP, CHVP/interferon-alfa 2a) have also demonstrated significant improvements in response rates, time dependent parameters as well as in overall survival (see Table 12).
Results from three other randomised studies using rituximab in combination with chemotherapy regimens other than CVP (CHOP, MCP, CHVP/interferon-alfa 2a) have also demonstrated significant improvements in response rates, time dependent parameters as well as in overall survival (see Table 12).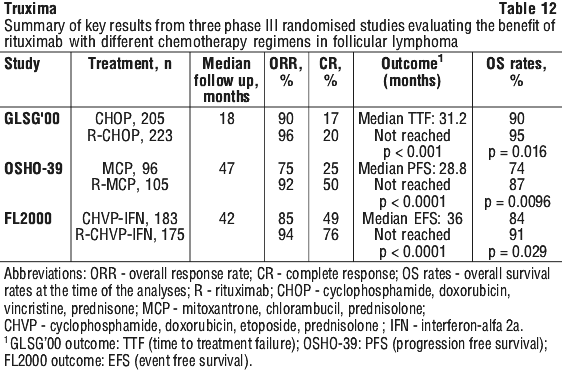
 For patients randomised to the maintenance phase of the trial, the median observation time was 28 months from maintenance randomisation. Maintenance treatment with rituximab led to a clinically relevant and statistically significant improvement in the primary endpoint, PFS, (time from maintenance randomisation to relapse, disease progression or death) when compared to observation alone (p < 0.0001 log-rank test).
For patients randomised to the maintenance phase of the trial, the median observation time was 28 months from maintenance randomisation. Maintenance treatment with rituximab led to a clinically relevant and statistically significant improvement in the primary endpoint, PFS, (time from maintenance randomisation to relapse, disease progression or death) when compared to observation alone (p < 0.0001 log-rank test).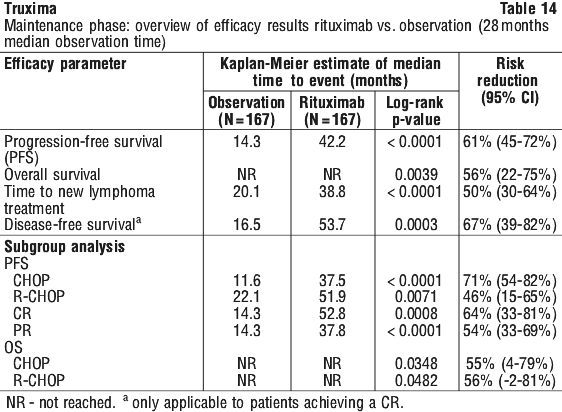 The benefit of rituximab maintenance treatment was confirmed in all subgroups analysed, regardless of induction regimen (CHOP or R-CHOP) or quality of response to induction treatment (CR or PR). Rituximab maintenance treatment significantly prolonged median PFS in patients responding to CHOP induction therapy (median PFS 37.5 months vs.11.6 months, p < 0.0001) as well as in those responding to R-CHOP induction (median PFS 51.9 months vs. 22.1 months, p = 0.0071). Although analysed subgroups were small, and the median survival had not been reached after an overall median observation period of 47.2 months, a clinically meaningful benefit in terms of overall survival was observed for patients receiving rituximab maintenance treatment when compared to observation, in the overall population.
The benefit of rituximab maintenance treatment was confirmed in all subgroups analysed, regardless of induction regimen (CHOP or R-CHOP) or quality of response to induction treatment (CR or PR). Rituximab maintenance treatment significantly prolonged median PFS in patients responding to CHOP induction therapy (median PFS 37.5 months vs.11.6 months, p < 0.0001) as well as in those responding to R-CHOP induction (median PFS 51.9 months vs. 22.1 months, p = 0.0071). Although analysed subgroups were small, and the median survival had not been reached after an overall median observation period of 47.2 months, a clinically meaningful benefit in terms of overall survival was observed for patients receiving rituximab maintenance treatment when compared to observation, in the overall population.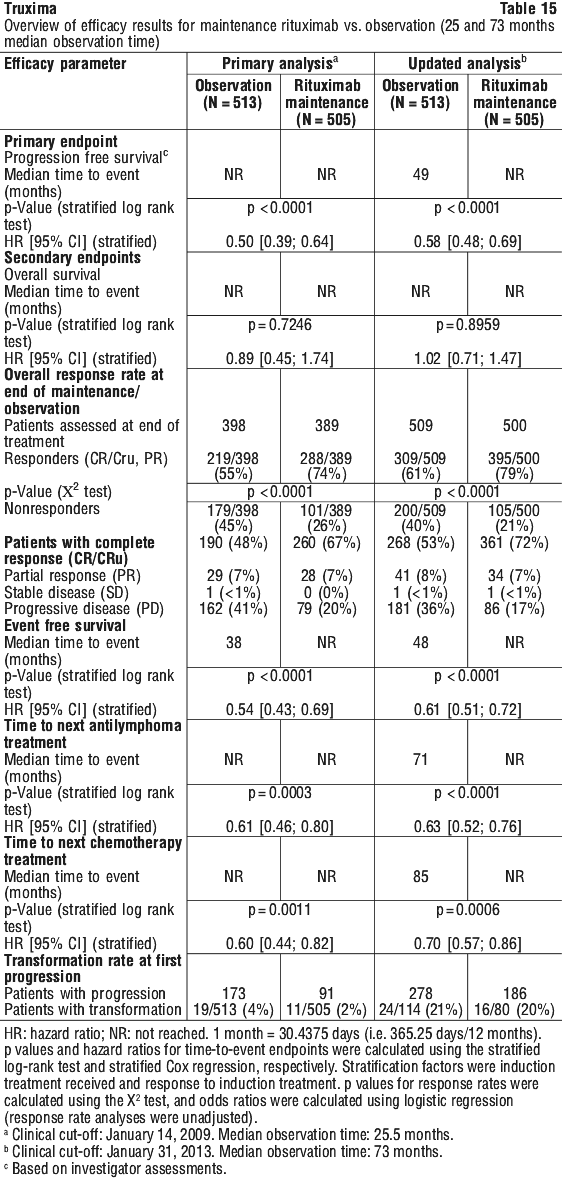 Rituximab maintenance treatment provided consistent benefit in all subgroups tested: gender (male, female), age (< 60 years, ≥ 60 years), FLIPI score (1, 2 or 3), induction therapy (R-CHOP, R-CVP or R-FCM) and regardless of the quality of response to induction treatment (CR or PR).
Rituximab maintenance treatment provided consistent benefit in all subgroups tested: gender (male, female), age (< 60 years, ≥ 60 years), FLIPI score (1, 2 or 3), induction therapy (R-CHOP, R-CVP or R-FCM) and regardless of the quality of response to induction treatment (CR or PR).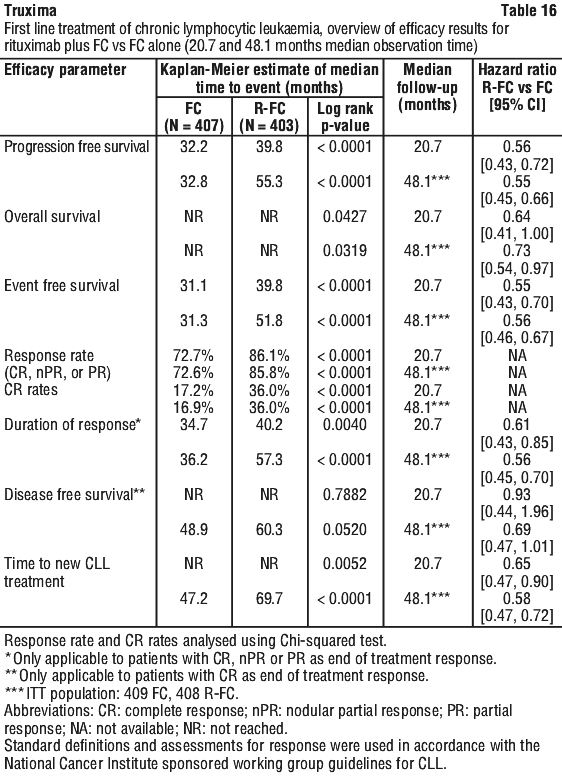
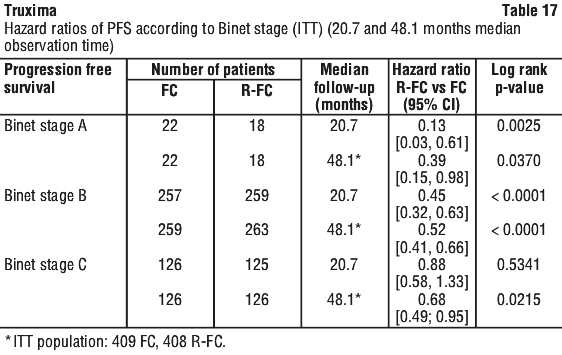 In a case series of 30 previously untreated patients with CLL, an overall response rate of 97% was achieved with rituximab in combination with fludarabine, cyclophosphamide and mitoxantrone (FCM). Survival was not reported. In another case series of 64 previously untreated patients with CLL, an overall response rate of 91% and a median PFS of 32.6 months were achieved with rituximab in combination with pentostatin and cyclophosphamide (PC).
In a case series of 30 previously untreated patients with CLL, an overall response rate of 97% was achieved with rituximab in combination with fludarabine, cyclophosphamide and mitoxantrone (FCM). Survival was not reported. In another case series of 64 previously untreated patients with CLL, an overall response rate of 91% and a median PFS of 32.6 months were achieved with rituximab in combination with pentostatin and cyclophosphamide (PC).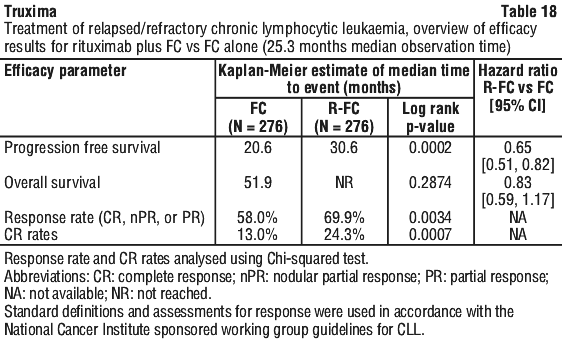 In relapsed/refractory CLL patients, response rates of 70% or greater have been reported in small studies of the following chemotherapy regimens with rituximab: FCM (fludarabine, cyclophosphamide, mitoxantrone), PC (pentostatin, cyclophosphamide), PCM (pentostatin, cyclophosphamide, mitoxantrone), CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone), bendamustine and cladribine.
In relapsed/refractory CLL patients, response rates of 70% or greater have been reported in small studies of the following chemotherapy regimens with rituximab: FCM (fludarabine, cyclophosphamide, mitoxantrone), PC (pentostatin, cyclophosphamide), PCM (pentostatin, cyclophosphamide, mitoxantrone), CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone), bendamustine and cladribine.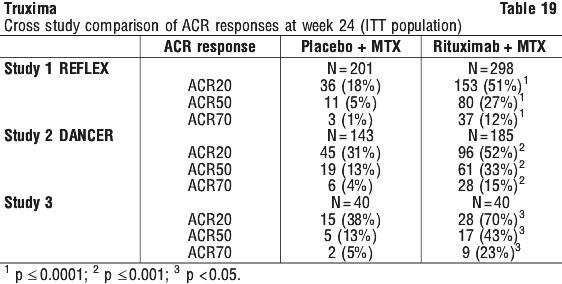 Rituximab + MTX treated patients had a significantly greater reduction in disease activity score (DAS28) than patients treated with MTX alone. A good to moderate EULAR response was achieved by significantly more rituximab + MTX treated patients compared to patients treated with MTX alone (see Table 20).
Rituximab + MTX treated patients had a significantly greater reduction in disease activity score (DAS28) than patients treated with MTX alone. A good to moderate EULAR response was achieved by significantly more rituximab + MTX treated patients compared to patients treated with MTX alone (see Table 20).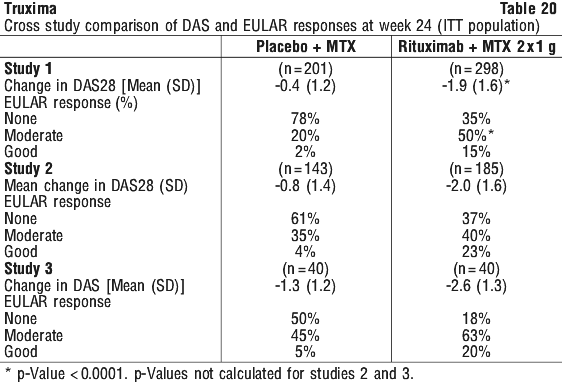
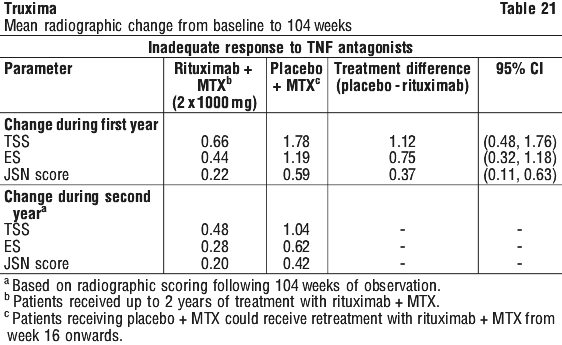 Following 2 years of treatment with rituximab + MTX, 57% of patients had no progression of structural damage. During the first year, 60% of rituximab + MTX treated patients had no progression, defined as a change in TSS of zero or less compared to baseline, compared to 46% of placebo + MTX treated patients. In their second year of treatment with rituximab + MTX, more patients had no progression than in the first year (68% vs. 60%), and 87% of the rituximab + MTX treated patients who had no progression in the first year also had no progression in the second year.
Following 2 years of treatment with rituximab + MTX, 57% of patients had no progression of structural damage. During the first year, 60% of rituximab + MTX treated patients had no progression, defined as a change in TSS of zero or less compared to baseline, compared to 46% of placebo + MTX treated patients. In their second year of treatment with rituximab + MTX, more patients had no progression than in the first year (68% vs. 60%), and 87% of the rituximab + MTX treated patients who had no progression in the first year also had no progression in the second year.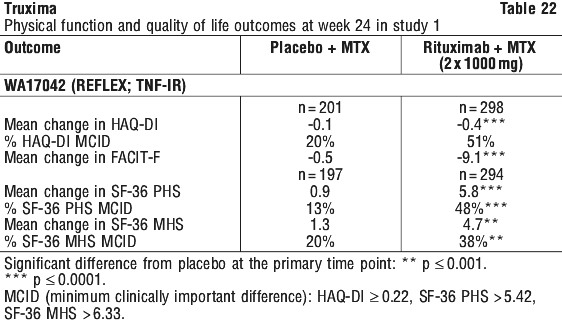 At week 24, in all three studies, the proportion of rituximab + MTX treated patients showing a clinically relevant improvement in HAQ-DI (defined as an individual total score decrease of > 0.25) was higher than among patients receiving MTX alone.
At week 24, in all three studies, the proportion of rituximab + MTX treated patients showing a clinically relevant improvement in HAQ-DI (defined as an individual total score decrease of > 0.25) was higher than among patients receiving MTX alone.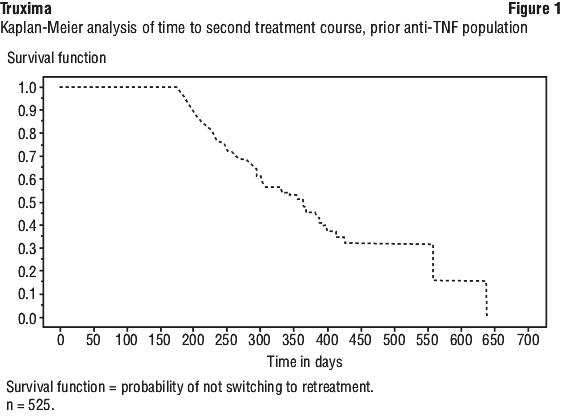
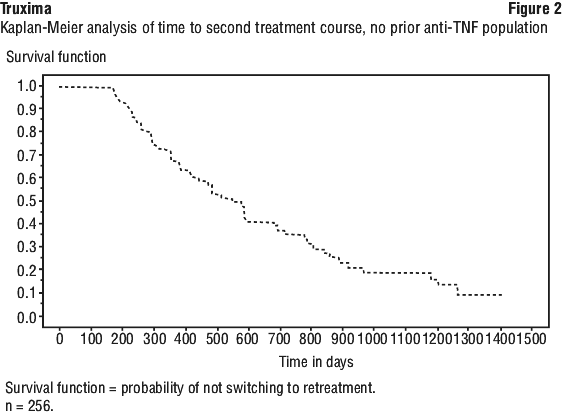
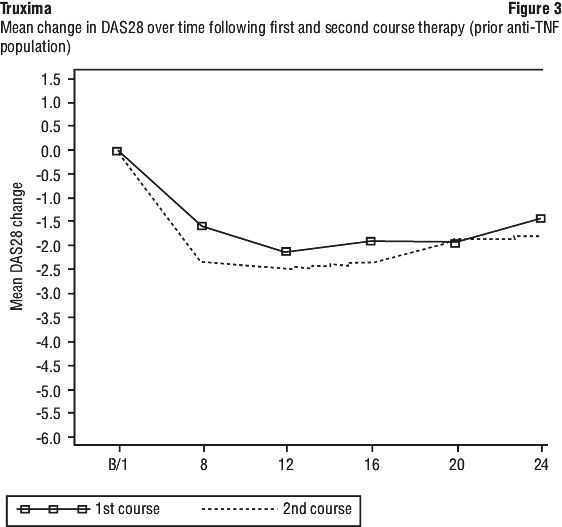
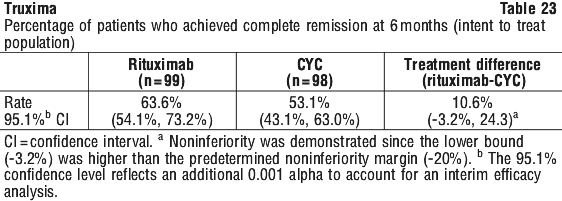
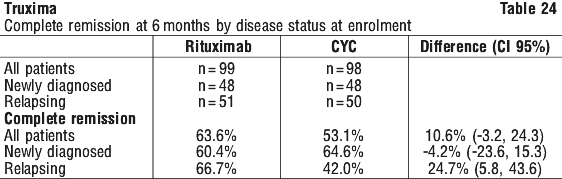
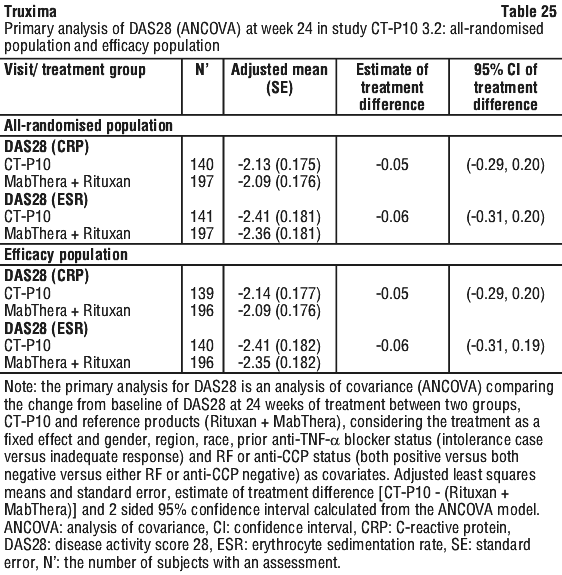
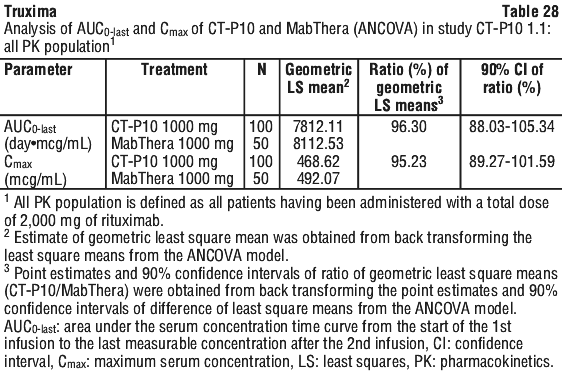
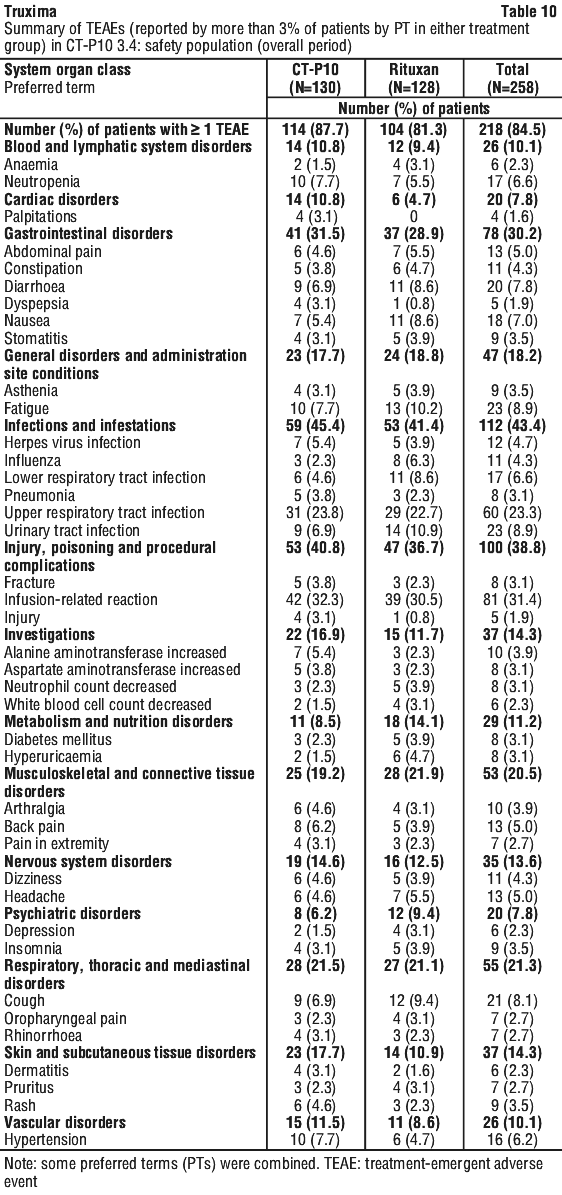
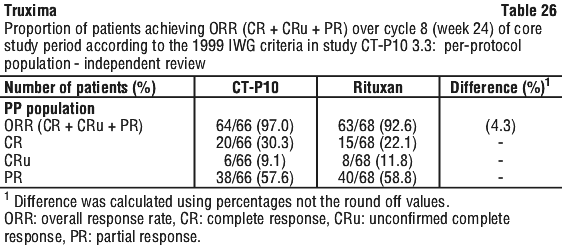 In Study CT-P10 3.3, efficacy in terms of overall response rate using the 1999 IWG criteria was evaluated over a 24 week period and efficacy was comparable between the CT-P10 and Rituxan groups.
In Study CT-P10 3.3, efficacy in terms of overall response rate using the 1999 IWG criteria was evaluated over a 24 week period and efficacy was comparable between the CT-P10 and Rituxan groups.