What is in this leaflet
Please read this leaflet carefully before you take any Tykerb tablets.
This leaflet answers some common questions about Tykerb. It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine.
You can also download the most up to date leaflet from www.novartis.com.au.
The updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the expected benefits of you taking Tykerb against the risks this medicine could have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What is Tykerb used for
Tykerb contains the active ingredient lapatinib, which belongs to a group of medicines called protein kinase inhibitors.
Tykerb is used in combination with other medicines to treat certain types of advanced or metastatic breast cancers. Breast cancer is caused by cells that divide abnormally in the breast. These cells may reach other tissues of the body at a later stage in a process called metastasis. Tykerb may slow or stop cancer cells from growing, or may kill them.
Advanced or metastatic breast cancers have spread beyond an original tumour. These types of breast cancers are more likely to grow in the presence of hormones, like oestrogen and progesterone.
Tykerb may be used with any of the following medicines:
- An aromatase inhibitor tablet to treat tumours that are hormone sensitive
- Capecitabine tablet to treat advanced or metastatic breast cancer whose tumours produce large amounts of a protein called human epidermal growth factor receptor-2 (HER2, also known as ErbB2) on the surface of the tumour cells.
- Paclitaxel infusion to treat metastatic breast cancer whose tumours produce large amounts of HER2 (ErbB2). These patients are unable to take another medicine containing trastuzumab.
Information about these other medicines is available in separate Consumer Medicine Information leaflets.
If you are taking any of these other medicines together with Tykerb, please read about the other medicine in the Consumer Medicine Information as well as this one carefully. If not included in the pack, the other medicine leaflets are available from your doctor or pharmacist.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed Tykerb for another reason.
This medicine is available only with a doctor's prescription.
It is not recommended for use in children and adolescents, under the age of 18 years.
Tykerb is not addictive.
Before you take Tykerb
When you must not take it
Do not take Tykerb if you have an allergy to:
- Lapatinib (active ingredient) or
- Any of the ingredients listed at the end of this leaflet.
(See "Ingredients".)
Some of the symptoms of an allergic reaction may include:
- Shortness of breath
- Wheezing or difficulty breathing
- Swelling of the face, lips, tongue or other parts of the body
- Rash, itching or hives on the skin
Do not take this medicine after the expiry date (EXP) printed on the pack has passed, or if the pack is torn or shows signs of tampering.
The expiry date refers to the last day of that month. If it is expired or damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor or nurse if you have allergies to any other medicines, foods, preservatives or dyes. Your doctor will want to know if you are prone to allergies.
Also tell your doctor if you have any of the following problems:
- Heart disorders, such as an irregular heartbeat.
- Lung disorders or problems breathing, including pain while breathing.
- Liver disorders.
Check with your doctor if you think any of these may apply to you. You may need extra tests to check that your heart and liver are working properly. Your doctor may decide to adjust your dose or stop treatment based on the results of these tests.
Taking other medicines
Tell your doctor or pharmacist if you are taking, have recently taken, or might take any other medicines. This includes herbal medicines and other medicines or complementary therapies that you may have bought without a prescription from your pharmacy, supermarket, or health food shop.
Keep a list of the medicines you take, so you can show it to your doctor, nurse or pharmacist when you get a new medicine. Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
Some medicines may affect the way Tykerb works or Tykerb may affect how other medicines work. These include:
- Erythromycin, ketoconazole, itraconazole, posaconazole, voriconazole, rifabutin, rifampicin, telithromycin (used to treat infections)
- Ritonavir, saquinavir (used to treat HIV)
- Cisapride (used to treat digestive system problems)
- Esomeprazole or other drugs that decrease stomach acidity (used to treat stomach ulcers or indigestion)
- Medicines used for sedation before surgery (anaesthesia), such as midazolam
- Quinidine, digoxin (used to treat heart problems)
- Verapamil (used to treat high blood pressure or angina)
- Rosuvastatin (used to treat high cholesterol)
- Repaglinide (used to treat diabetes)
- Phenytoin, carbamazepine (used to treat seizures)
- Pimozide (used to treat mental health problems)
- Nefazodone (used to treat depression)
- St John's Wort (a herb extract used to treat depression)
- Cyclosporin (used to prevent the rejection of transplanted organs)
- Topotecan, paclitaxel, irinotecan, docetaxel (used to treat cancer).
Tell your doctor if you are taking any of these.
Taking Tykerb with food and drink
Tykerb is affected by food intake and must be taken on an empty stomach, at least 1 hour before or 1 hour after eating.
You should not drink grapefruit juice or eat grapefruit during your treatment with Tykerb. It may make this medicine less effective and possibly increase the chance of side effects.
Pregnancy
If you are pregnant, think you could be pregnant, or are planning to have a baby, ask your doctor or healthcare provider for advice before taking Tykerb.
Tykerb may harm your unborn baby.
You should avoid becoming pregnant while taking Tykerb.
Use a reliable method of birth control (contraception) during treatment and for at least 5 days after stopping Tykerb.
Ask your doctor about effective contraception options. Your doctor may recommend that you don't take Tykerb while you are pregnant.
If you become pregnant or think you are pregnant, tell your doctor or healthcare provider right away.
Breast feeding
Do not breast-feed while taking Tykerb and for 5 days after the last dose as it may harm your baby. Ask your doctor for advice.
Tell your doctor if you are breast-feeding. Your doctor will discuss with you the risks of taking Tykerb during breast-feeding.
Driving and using machines
Tykerb can cause tiredness and may make you unfit to drive.
Do not drive or operate machinery unless you're feeling well and are sure that you are not affected by tiredness.
How to take Tykerb
Follow all directions given to you by your doctor or pharmacist. The directions may differ from the information contained in this leaflet.
If you do not understand the instructions on the label ask your doctor or pharmacist for help.
Your doctor will tell you exactly how many Tykerb tablets that you should take. Depending on your response to Tykerb or if you have heart, lung or liver problems, or experience serious episodes of diarrhoea or skin reactions during treatment with Tykerb, your doctor may prescribe a lower dose or temporarily stop treatment.
When to take Tykerb
It is important that you take Tykerb tablets either at least one hour before or at least one hour after food.
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How much to take
Your doctor will tell you the dose of capecitabine, paclitaxel or aromatase inhibitor that you should take and when you should take it.
If you are aged 65 years or above you can use Tykerb at the same dose as for other adults.
In combination with capecitabine:
The usual dose is 5 (five) Tykerb tablets (a total dose of 1250 mg) taken once a day.
In combination with paclitaxel:
The usual dose is 6 (six) Tykerb tablets (a total dose of 1500 mg) taken once a day.
In combination with an aromatase inhibitor:
The usual dose is 6 (six) Tykerb tablets (a total dose of 1500 mg) taken once a day.
How to take it
Tykerb tablets should be swallowed with a whole with water. Tykerb film-coated tablets should not be chewed, crushed or split prior to swallowing.
If you have to take another medicine to treat your breast cancer in addition to Tykerb, follow your doctor's instructions on how to take that medicine
If you forget to take Tykerb
Take the next dose at the scheduled time. Then go back to taking your medicine as you would normally.
Do not take a double dose to make up for a missed dose. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your doctor, nurse or pharmacist for some hints.
How long to take it for
Keep taking Tykerb for as long as your doctor recommends. Don't stop unless your doctor advises you to. Stopping your treatment with Tykerb is likely to cause your condition to become worse.
This is a long-term treatment that may continue for months or years. Your doctor will regularly monitor your condition to check that the treatment is having the desired effect.
If you have questions about how long to take Tykerb, talk to your doctor or healthcare provider.
If you take more Tykerb than you should (Overdose)
If you have taken too much Tykerb, or if somebody else accidentally takes your medicine, immediately telephone your doctor or Poisons Information Centre (In Australia call 131126) for advice, or go to Accident and Emergency at the nearest hospital.
Do this even if there are no signs of discomfort or poisoning. You/they may need urgent medical attention.
Take the Tykerb pack with you. You may need to show medical staff your pack.
While you are taking Tykerb
Things you must do
Take Tykerb for as long as your doctor recommends.
Don't stop taking this medicine unless your doctor advises you to.
Tell your doctor if, for any reason, you have not taken your medicine exactly as directed. Otherwise, your doctor may think that it was not working as it should and change your treatment unnecessarily.
Keep all of your doctor's appointments so that your progress can be checked. Your heart, lung and liver function will be regularly checked before and during treatment with Tykerb.
If you are a woman that could become pregnant, you must use an effective contraceptive to prevent pregnancy during treatment with Tykerb and for 5 days after the last dose of Tykerb.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Tykerb.
Tell any doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you have any further questions on the use of this medicine, ask your doctor or pharmacist.
Things you must not do
Do not drink grapefruit juice while you are being treated with Tykerb. (See "Taking Tykerb with food and drink.")
Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
Do not use Tykerb to treat any other complaints unless your doctor says to.
Do not stop taking Tykerb unless your doctor advises you to.
Do not dispose of medicines in wastewater or household rubbish. This will help to protect the environment.
Possible side effects
As with all medicines, patients treated with Tykerb alone or in combination with capecitabine, paclitaxel, or an aromatase inhibitor may experience side effects, although not everybody gets them.
Do not be alarmed by these possible side effects. You may not experience any of them. If they occur, they are most likely to be minor and temporary. However, some may be serious and need medical attention.
Check with your doctor as soon as possible if you think you are experiencing any side effects or allergic reactions due to taking Tykerb, even if the problem is not listed below.
These side effects have occurred with Tykerb alone, or in combination with capecitabine, paclitaxel or letrozole (an aromatase inhibitor).
Serious side effects
STOP taking Tykerb and seek medical help immediately if you experience any of the following side effects.
Very common serious side effects
These may affect more than 1 in 10 people:
- Fever, sore throat, frequent infections as signs of low level of white blood cells (leukopenia)
- Pale skin, weakness, frequent infections with fever, chills and sore throat as signs of low level of red blood cells (anaemia)
Common serious side effects
These may affect up to 1 in 10 people:
- Irregular heartbeat and shortness of breath (decreased left ventricular ejection fraction)
Uncommon serious side effects
These may affect up to 1 in 100 people:
- Itching, yellow eyes or skin (jaundice), dark urine or pain or discomfort in the right upper area of the stomach (hepatotoxicity or hyperbilirubinemia)
- Cough or shortness of breath (interstitial lung disease and/or pneumonitis).
Rare serious side effects
These may affect up to 1 in 1000 people:
- Skin rash (including itchy, bumpy rash), skin reddening, hives, unusual wheezing or coughing or difficulty breathing, swollen eyelids, lips, face or tongue, blue discoloration of the lips, tongue, or skin, pain in muscles or joints, light-headedness, dizziness, loss of consciousness (passing out), hypotension (signs of severe allergic reactions).
Frequency unknown serious side effects
The frequency of these side effects is not known (events from spontaneous reports):
- Irregular heart-beat (ventricular arrhythmia/Torsade de Pointes)
- Change in the electrical activity of the heart (QT interval in the electrocardiogram prolonged)
- Rash, red skin, blistering of the lips, eyes or mouth, skin peeling, fever or any combination of these (Stevens-Johnson syndrome, toxic epidermal necrolysis).
Other possible side effects
Other side effects include those listed below. If these side effects are severe, please tell your doctor, pharmacist or healthcare provider immediately.
Very common
These may affect more than 1 in 10 people:
- Diarrhoea (can be life-threatening if severe); report any serious change in bowel patterns, such as loose stool immediately.
- A skin reaction or pain on the palms of the hands or soles of the feet, including tingling, numbness, pain, swelling or reddening (palmar-plantar erythrodysesthesia or hand-foot syndrome)
- Muscle pain
- Numbness, tingling or weakness of the arms and legs
- Loss of appetite (anorexia)
- Indigestion or stomach pain (dyspepsia)
- Feeling or being sick (nausea or vomiting)
- Constipation
- Tiredness (fatigue)
- Unusual hair loss or thinning (alopecia)
- Nose bleed (epistaxis)
- Sore mouth or mouth ulcers (mucosal inflammation)
- Trouble sleeping (insomnia)
- Back pain
- Pain in extremity
- Dry skin
- Rash.
Common
These may affect up to 1 in 10 people:
- Headache
- Nail disorders - such as a tender infection and swelling of the bottom part of the nail (cuticles).
- Deep cracks on the skin or chapped skin (skin fissures)
If you notice any side effects not listed, please inform your doctor or healthcare provider.
After taking Tykerb
Storage
Keep this medicine where children cannot see or reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Store the tablets in a cool place below 30°C.
Do not leave this medicine in a car, on a window sill or in a bathroom. Heat and dampness can destroy some medicines.
Keep Tykerb in its original container until it is time to take it.
Disposal
If your doctor tells you to stop taking this medicine or if you have any unwanted medicines, ask your pharmacist what to do with them.
Product description
What Tykerb looks like
Tykerb is supplied in plastic bottle packs containing 70 tablets.
250 mg tablets
Tykerb tablets are oval, rounded on both sides, yellow film-coated, and with GS XJG imprinted on one side.
Ingredients
Each Tykerb tablet contains the active ingredient lapatinib ditosilate monohydrate, equivalent to 250 mg of lapatinib.
Tykerb tablets also contain the following ingredients:
- Microcrystalline cellulose (E460)
- Povidone (E1201)
- Sodium starch glycollate type A
- Magnesium stearate (E572) (vegetable origin)
- Hypromellose (E464)
- Titanium dioxide (E171)
- Macrogol (E1521)
- Polysorbate (E433)
- Iron oxide yellow (E172)
- Iron oxide red (E172).
Tykerb tablets do not contain lactose, sucrose, tartrazine or any other azo dyes.
Sponsor
Tykerb is supplied in Australia by:
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road, Macquarie Park
NSW 2113 Australia
Telephone 1 800 671 203
www.novartis.com.au
® = Registered Trademark
This leaflet was prepared in
May 2022
Australian Registration Number:
AUST R 185997: Tykerb 250 mg bottle pack
Internal document code:
tyk270522c based on PI tyk270522i
Published by MIMS July 2022
 No age based differences in the safety or efficacy of these regimens were observed. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Greater sensitivity of elderly individuals cannot be ruled out.
No age based differences in the safety or efficacy of these regimens were observed. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Greater sensitivity of elderly individuals cannot be ruled out.
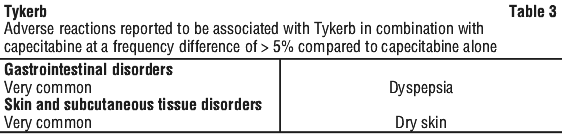






 In the Tykerb plus letrozole treatment group, the most commonly observed study medication related serious adverse events were decreased left ventricular ejection fraction (LVEF) (2% of patients, compared to 1% for letrozole plus placebo) and diarrhoea (2% of patients, compared to < 1% for letrozole plus placebo). Other study medication related serious adverse events, including skin rash, hepatotoxicity and pneumonitis, were observed in < 1% of patients. The most common adverse events leading to discontinuation of treatment in the Tykerb plus letrozole treatment group were diarrhoea (4%) and vomiting (2%) (see Table 10).
In the Tykerb plus letrozole treatment group, the most commonly observed study medication related serious adverse events were decreased left ventricular ejection fraction (LVEF) (2% of patients, compared to 1% for letrozole plus placebo) and diarrhoea (2% of patients, compared to < 1% for letrozole plus placebo). Other study medication related serious adverse events, including skin rash, hepatotoxicity and pneumonitis, were observed in < 1% of patients. The most common adverse events leading to discontinuation of treatment in the Tykerb plus letrozole treatment group were diarrhoea (4%) and vomiting (2%) (see Table 10).
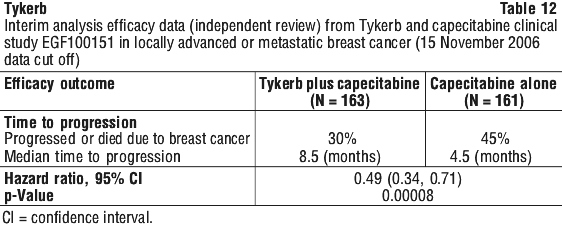 The response rate (complete or partial response) independently assessed was 22% in the Tykerb plus capecitabine group compared with 14% in the capecitabine group (p = 0.091); similar results were observed for the clinical benefit response rate (complete response + partial response + stable disease for at least 6 months), which was 27% vs 18% (p = 0.069) in the combination versus the monotherapy arm, respectively.
The response rate (complete or partial response) independently assessed was 22% in the Tykerb plus capecitabine group compared with 14% in the capecitabine group (p = 0.091); similar results were observed for the clinical benefit response rate (complete response + partial response + stable disease for at least 6 months), which was 27% vs 18% (p = 0.069) in the combination versus the monotherapy arm, respectively. An independent data monitoring committee (IDMC) initially reviewed the results of the interim analysis (which included data from 321 of the 324 patients) and recommended that further enrollment into the study be halted due to a statistically significant and clinically relevant increase in TTP for the combination of Tykerb and capecitabine over capecitabine alone, which crossed a predefined statistical stopping boundary for superiority. At the time enrollment was halted (3 April, 2006), a total of 399 patients had been randomised to study treatment.
An independent data monitoring committee (IDMC) initially reviewed the results of the interim analysis (which included data from 321 of the 324 patients) and recommended that further enrollment into the study be halted due to a statistically significant and clinically relevant increase in TTP for the combination of Tykerb and capecitabine over capecitabine alone, which crossed a predefined statistical stopping boundary for superiority. At the time enrollment was halted (3 April, 2006), a total of 399 patients had been randomised to study treatment.
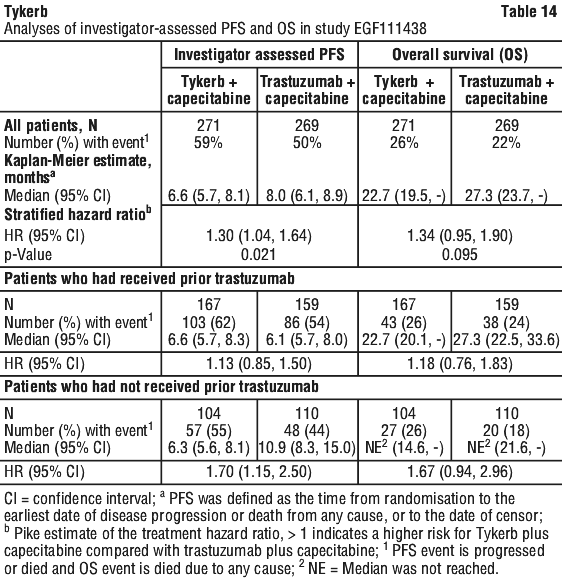






 In the intent to treat (ITT) population, investigator determined PFS was greater between the two treatment arms (see Table 19). Although statistically significant, the difference was not considered clinically relevant.
In the intent to treat (ITT) population, investigator determined PFS was greater between the two treatment arms (see Table 19). Although statistically significant, the difference was not considered clinically relevant.
