1 Name of Medicine
Faricimab.
2 Qualitative and Quantitative Composition
Each vial of 0.24 mL contains 28.8 mg of faricimab at a concentration of 120 mg/mL. Each prefilled syringe of 0.175 mL contains 21 mg of faricimab at a concentration of 120 mg/mL. This provides a usable amount to deliver a single dose of 0.05 mL solution containing 6 mg of faricimab.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection for intravitreal use.
Clear to opalescent, colourless to brownish-yellow liquid.
4.1 Therapeutic Indications
Vabysmo is indicated for the treatment of:
neovascular (wet) age-related macular degeneration (nAMD);
diabetic macular oedema (DMO);
macular oedema secondary to retinal vein occlusion (RVO).
4.2 Dose and Method of Administration
For intravitreal injection only. Vabysmo must be administered by a qualified ophthalmologist experienced in intravitreal injections. Each vial or pre-filled syringe should only be used for the treatment of a single eye.
Dosage.
Neovascular (wet) age-related macular degeneration (nAMD).
The recommended dose for Vabysmo is 6 mg (0.05 mL) administered by intravitreal injection every 4 weeks for the first 4 doses. Thereafter, an assessment of disease activity based on anatomic and/or visual outcomes is recommended 20 - 24 weeks after treatment initiation so that treatment can be individualised ("treat-and-extend" approach). Based on these assessments, in patients without disease activity, administration of Vabysmo every 16 weeks should be considered. In patients with disease activity, treatment every 8 weeks or 12 weeks should be considered. If anatomic and/or visual outcomes change, the treatment interval should be adjusted accordingly, and interval reduction should be implemented if anatomic and/or visual outcomes deteriorate (see Section 5.1). There is limited safety data on treatment intervals of 8 weeks or less between injections (see Section 4.4).
Monitoring between dosing visits should be scheduled based on the patient's status and at the ophthalmologist's discretion.
Diabetic macular oedema (DMO).
The recommended dose for Vabysmo is 6 mg (0.05 mL) administered by intravitreal injection every 4 weeks for the first 4 doses. Thereafter, treatment may be individualised using a treat-and-extend approach following an assessment of the individual patient's anatomic and/or visual outcomes. Following the outcome of this assessment, the dosing interval may remain at every 4 weeks, or may be extended in 4 week increments up to every 16 weeks. If anatomic or visual outcomes change, the treatment interval should be adjusted accordingly (see Section 5.1).
Continued monitoring of disease activity and individualisation of dosing is recommended. Monitoring between the dosing visits should be scheduled based on the patient's status and at the ophthalmologist's clinical judgement.
Macular oedema secondary to retinal vein occlusion (RVO).
The recommended dose for Vabysmo is 6 mg (0.05 mL solution) administered by intravitreal injection every 4 weeks; 3 or more consecutive 4 weekly injections may be needed.
Thereafter, treatment may be individualised using a treat-and-extend approach following an assessment of the individual patient's anatomic and/or visual outcomes. The dosing interval may be extended, in increments of up to 4 weeks. If anatomic and/or visual outcomes change, the treatment interval should be adjusted accordingly, and interval reduction should be implemented if anatomic and/or visual outcomes deteriorate (see Section 5.1). Treatment intervals longer than 16 weeks between injections have not been studied.
Monitoring between dosing visits should be scheduled based on the patient's status and at the ophthalmologist's clinical judgement.
Duration of treatment.
Vabysmo can be used for long-term treatment, if clinically indicated. If visual and/or anatomic outcomes indicate that the patient is not benefitting from continued treatment, Vabysmo should be discontinued.
Delayed or missed dose.
If a dose is delayed or missed, the patient should return to be assessed by the ophthalmologist at the next available visit and continue dosing depending on the ophthalmologist's discretion.
Dose modification.
No dose modifications of Vabysmo are recommended.
Special populations.
Paediatric use.
The safety and efficacy of Vabysmo in pediatric patients have not been established.
Geriatric use.
No dose adjustment is required in patients ≥ 65 years of age (see Section 5.2).
Renal impairment.
No dose adjustment is required in patients with renal impairment (see Section 5.2).
Hepatic impairment.
No dose adjustment is required in patients with hepatic impairment (see Section 5.2).
Method of administration.
Single-use vial for intravitreal use in one patient only. Discard any residue.
Immediately following the intravitreal injection, patients should be monitored for elevation in intraocular pressure. Appropriate monitoring may consist of a check for perfusion of the optic nerve head or tonometry. If required, sterile equipment for paracentesis should be available.
Following intravitreal injection patients should be instructed to report any symptoms suggestive of endophthalmitis (e.g. vision loss, eye pain, redness of the eye, photophobia, blurring of vision) without delay.
Instructions for use and handling of Vabysmo.
Vabysmo should be inspected visually for particulate matter and discoloration prior to administration. Vabysmo is a clear to opalescent and colourless to brownish-yellow liquid solution. Do not use if particles, cloudiness or discolouration are visible.
Vabysmo vial. Allow the Vabysmo vial to reach room temperature (20°C to 25°C) before use. Keep the vial in the original carton to protect from light. The unopened Vabysmo vial may be stored at room temperature (25°C) for up to 24 hours.
Preparation of the intravitreal injection.
Use aseptic technique to carry out the preparation of the intravitreal injection.
To ensure all liquid settles at the bottom of the vial, place the vial upright on a flat surface (for about 1 minute) after removal from packaging. Gently tap the vial with your finger, as liquid may stick to the top of the vial.
Remove the flip-off cap from the vial and disinfect the vial septum.
Firmly attach the included 18 G x 1½ inch transfer filter needle onto a 1 mL luer lock syringe.
Push the transfer filter needle into the centre of the vial septum until the needle is completely inserted into the vial. Tilt the vial slightly so that the needle touches the bottom edge of the vial.
Hold the vial slightly inclined and slowly withdraw all the liquid from the vial. The bevel of the transfer filter needle must be kept submerged in the liquid, to avoid introduction of air.
Ensure that the plunger rod is drawn sufficiently back when emptying the vial, in order to completely empty the transfer filter needle.
Disconnect the transfer filter needle from the syringe and appropriately dispose of it. Do not use the transfer filter needle for the intravitreal injection.
Aseptically and firmly attach a 30 G x ½ inch injection needle onto the luer lock syringe.
Hold the syringe with the needle pointing up to check for air bubbles. If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top.
Carefully expel the air from the syringe and needle and slowly depress the plunger to align the rubber stopper tip to the 0.05 mL dose mark.
Ensure that the injection is given immediately after preparation of the dose.
Inject slowly until the rubber stopper reaches the end of the syringe to deliver the volume of 0.05 mL. Confirm delivery of the full dose by checking that the rubber stopper has reached the end of the syringe barrel.
Dispose of any waste material or unused medicinal product.
Vabysmo pre-filled syringe. Allow the Vabysmo pre-filled syringe to reach room temperature (20°C to 25°C) before use. Keep the sealed tray in the original carton to protect from light. The unopened Vabysmo pre-filled syringe may be stored at room temperature (25°C) for up to 24 hours.
Preparation of the intravitreal injection.
Use aseptic technique to carry out the preparation of the intravitreal injection.
Peel the lid off the syringe tray and remove the pre-filled syringe.
Hold the syringe by the white collar; snap off the syringe cap. Do not twist off the cap.
Remove the filter needle from its packaging.
Firmly attach the injection filter needle onto the luer lock syringe.
Hold the syringe with the injection filter needle pointing up. Check the syringe for air bubbles.
If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top.
Hold the syringe at eye level and slowly push the plunger rod until the lower edge of the rubber stopper's dome is aligned with the 0.05 mL dose mark. This will expel the air and the excess solution and set the dose to 0.05 mL.
Ensure that the injection is given immediately after preparation of the dose.
Inject slowly until the rubber stopper reaches the end of the syringe to deliver the volume of 0.05 mL.
Do not recap or detach the injection filter needle from the syringe. Dispose of any waste material or unused medicinal product.4.3 Contraindications
Vabysmo is contraindicated in patients with:
A known hypersensitivity to faricimab or any of the excipients listed in Section 6.1. Hypersensitivity reactions may manifest as rash, pruritus, urticaria, erythema, or severe intraocular inflammation.
Ocular or periocular infections.
Active intraocular inflammation.
4.4 Special Warnings and Precautions for Use
Intravitreal injection-related reactions.
Intravitreal injections, including those with Vabysmo have been associated with endophthalmitis, intraocular inflammation, rhegmatogenous retinal detachment, retinal tear and iatrogenic traumatic cataract. Proper aseptic injection techniques must always be used when administering Vabysmo. Patients should be instructed to report any symptoms, such as pain, loss of vision, photophobia, blurred vision, floaters, or redness, suggestive of endophthalmitis or any of the above-mentioned events without delay, to permit prompt and appropriate management.
Retinal vasculitis and/or retinal occlusive vasculitis.
Retinal vasculitis and/or retinal occlusive vasculitis have been reported with the use of Vabysmo in the post-marketing setting. Discontinue treatment with Vabysmo in patients who develop these events. Patients should be instructed to report any change in vision without delay (see Section 4.8).
Intraocular pressure increase.
Transient increases in intraocular pressure (IOP) have been seen within 60 minutes of intravitreal injection, including those with Vabysmo. Vabysmo has not been studied in patients with poorly controlled glaucoma. Special precaution is needed in patients with poorly controlled glaucoma (do not inject Vabysmo while the IOP is ≥ 30 mmHg). In all cases, both the IOP and perfusion of the optic nerve head and/or vision must be monitored and managed appropriately.
Systemic effects.
Systemic adverse events including arterial thromboembolic events, such as stroke and myocardial infarction, have been reported following intravitreal injection of vascular endothelial growth factor (VEGF) inhibitors and there is a theoretical risk that these may be related to VEGF inhibition. There is limited safety data in patients with a prior history of stroke or transient ischaemic attack or myocardial infarction.
Immunogenicity.
As with all therapeutic proteins, there is the potential for immune response to Vabysmo. Patients should be instructed to report any signs or symptoms of intraocular inflammation such as vision loss, eye pain, increased sensitivity to light, floaters or worsening eye redness, which might be a clinical sign attributable to hypersensitivity (see Section 4.8).
Bilateral treatment.
The safety and efficacy of Vabysmo administered in both eyes concurrently have not been studied.
Concomitant use of other anti-VEGF.
There are no data available on the concomitant use of Vabysmo with anti-VEGF medicinal products in the same eye.
Withholding treatment.
Treatment should be withheld in patients with:
Rhegmatogenous retinal detachment, stage 3 or 4 macular holes, retinal break; treatment should not be resumed until an adequate repair has been performed.
Treatment related decrease in Best Corrected Visual Acuity (BCVA) of ≥ 30 letters compared with the last assessment of visual acuity; treatment should not be resumed earlier than the next scheduled treatment.
Performed or planned intraocular surgery within the previous or next 28 days guided by the ophthalmologist's clinical judgement; treatment should not be resumed earlier than the next scheduled treatment.
Retinal pigment epithelial tear.
Retinal pigment epithelial (RPE) tear is a complication of pigment epithelial detachment (PED) in patients with nAMD. Risk factors associated with the development of a retinal pigment epithelial tear after anti-VEGF therapy for nAMD include a large and/or high pigment epithelial detachment. When initiating Vabysmo therapy, caution should be used in patients with these risk factors for retinal pigment epithelial tears.
Populations with limited data.
There is only limited experience in the treatment of nAMD patients ≥ 85 years, and DMO patients with type I diabetes, patients with HbA1c over 10%, patients with high-risk proliferative diabetic retinopathy (DR), high blood pressure (≥ 140/90 mmHg) and vascular disease, sustained dosing intervals shorter than every 8 weeks (Q8W), or nAMD, DMO and RVO patients with active systemic infections. There is limited safety information on sustained dosing intervals of 8 weeks or less and these may be associated with a higher risk of ocular and systemic adverse reactions, including serious adverse reactions. There is also no experience of treatment with Vabysmo in diabetic and RVO patients with uncontrolled hypertension. This lack of information should be considered by the ophthalmologist when treating such patients.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No drug-drug interaction studies have been performed with Vabysmo.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No reproductive or fertility studies have been conducted. No effects on reproductive organs in male or female animal were observed in a 6-month cynomolgus monkey study at faricimab doses of up to 3 mg/eye (10 times the clinical exposures based on AUC). VEGF inhibition has been shown to affect follicular development, corpus luteum function and fertility. Based on the mechanism of action of VEGF and Ang-2 inhibitors, there is a potential risk to female reproductive capacity however the risk is considered low due to the low systemic exposure after ocular administration.
Women of childbearing potential.
Women of childbearing potential should use contraception during treatment with Vabysmo and for at least 3 months following the last dose of Vabysmo.
(Category D)
There is no data from the use of Vabysmo in pregnant women.
In an embryofetal development study in pregnant cynomolgus monkeys, faricimab given intravenously throughout the period of organogenesis did not adversely affect pregnancy or fetal development at doses up to 3 mg/kg IV (523 times the clinical exposures based on the Cmax at the MRHD of a single 6 mg intravitreal dose).
VEGF inhibition has been shown to cause malformations, embryofetal resorption and decreased fetal weight. No dedicated studies addressing the effects of Ang-2 inhibition on pregnancy are available. Based on nonclinical information Ang-2 inhibition may lead to effects comparable to VEGF inhibition. Systemic exposures after ocular administration of Vabysmo is very low.
It is not known whether faricimab can cross the placenta or cause harm to the fetus when administered to pregnant women. Based on the mechanism of action of VEGF and Ang-2 inhibitors, there is a potential risk to female reproductive capacity, and to embryo-fetal development. Although the systemic exposure after ocular administration is very low, Vabysmo should not be used during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus.
The safe use of Vabysmo during labour and delivery has not been established.
It is not known whether faricimab is excreted in human breast milk. Animal studies have not been conducted to assess the impact of faricimab on milk production or its presence in breast milk. Because many drugs are excreted in human milk with the potential for absorption and harm to infant growth and development exists, caution should be exercised when Vabysmo is administered to a nursing woman. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Vabysmo and any potential adverse effects on the breastfed child from Vabysmo.4.7 Effects on Ability to Drive and Use Machines
Vabysmo may have a minor influence on the ability to drive and use machines due to possible temporary visual disturbances following the intravitreal injection and the associated eye examination. Patients should not drive or use machines until visual function has recovered sufficiently.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
A total of 4,489 patients constituted the safety population in the six Phase III clinical studies for two years (2,567 Vabysmo treated patients; 664 in nAMD, 1,262 in DMO and 641 in RVO).
The most serious adverse reactions were uveitis (0.5%), endophthalmitis (0.4%), vitritis (0.4%), retinal tear (0.2%), rhegmatogenous retinal detachment (0.1%) and traumatic cataract (< 0.1%).
The most frequently reported adverse reactions in patients treated with Vabysmo were cataract (9.7%), conjunctival haemorrhage (6.7%), vitreous detachment (4.2%), intraocular pressure increased (3.5%), vitreous floaters (3.5%), eye pain (2.5%) and retinal pigment epithelial tear (nAMD only) (2.9%).
Tabulated summary of adverse reactions from clinical trials.
The safety data described below includes all adverse reactions from the pooled data across six Phase III clinical studies in the indications nAMD, DMO and RVO, with a reasonable possibility of causality attribution to the injection procedure or medicinal product.
The adverse reactions are listed according to the MedDRA system organ class and ranked by frequency using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000). See Table 1.

Description of selected adverse drug reactions from clinical trials.
There is a theoretical risk of arterial thromboembolic events, including stroke and myocardial infarction, following intravitreal use of VEGF inhibitors. A low incidence rate of arterial thromboembolic events was observed in the Vabysmo clinical trials in patients with nAMD, DMO and RVO. Across indications no notable difference between the groups treated with Vabysmo and the comparator were observed.
Immunogenicity.
There is a potential for an immune response in patients treated with Vabysmo (see Section 4.4). After dosing with Vabysmo for up to 112 (nAMD), 100 (DMO) and 72 (RVO) weeks, treatment-emergent anti-faricimab antibodies were detected in approximately 13.8% and 9.6% and 14.4% of patients with nAMD, DMO and RVO randomised to Vabysmo, respectively. The clinical significance of anti-faricimab antibodies on safety is unclear at this time. The incidence of intraocular inflammation in anti-faricimab antibody positive patients was 12/98 (12.2%; nAMD), 15/128 (11.7%; DMO) and 9/95 (9.5%; RVO), and in anti-faricimab antibody negative patients was 8/562 (1.4%; nAMD), 5/1124 (0.4%; DMO) and 10/543 (1.8%; RVO). The incidence of serious ocular adverse reactions in anti-faricimab antibody positive patients was 6/98 (6.1%; nAMD), 14/128 (10.9%; DMO) and 7/95 (7.4%; RVO), and in anti-faricimab antibody negative patients was 23/562 (4.1%; nAMD), 45/1124 (4.0%; DMO) and 34/543 (6.3%; RVO). Anti-faricimab antibodies were not associated with an impact on clinical efficacy or systemic pharmacokinetics.
Retinal pigment epithelial tear.
Retinal pigment epithelial (RPE) tear is a complication of pigment epithelial detachment (PED) in patients with nAMD. RPE tears are common in nAMD patients with PED, treated with IVT anti-VEGF agents including faricimab. There was a higher rate of RPE tear in the faricimab group (2.9%) compared to aflibercept group (1.4%). The majority of events were mild to moderate, without impact to vision and occurred during the loading phase.
Post-marketing experience.
Rare cases of retinal vasculitis and/or retinal occlusive vasculitis have been spontaneously reported in the post-marketing setting. Retinal vasculitis and retinal occlusive vasculitis have also been reported in patients treated with intravitreal (IVT) therapies.
Eye disorders.
Retinal vasculitis, retinal occlusive vasculitis.
Reporting of suspected adverse reactions.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Doses higher than the recommended dosing regimen have not been studied. Overdosing with greater than recommended injection volume may increase intraocular pressure.
In the event of an overdose, IOP should be monitored and, if deemed necessary by the treating ophthalmologist, appropriate treatment should be initiated.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Ophthalmologicals, other ocular vascular disorder agents. ATC code: S01LA09.
Mechanism of action.
Faricimab is a humanised bispecific immunoglobulin G1 (IgG1) antibody that acts through inhibition of two distinct pathways by neutralisation of both Ang-2 and vascular endothelial growth factor A (VEGF-A).
Ang-2 causes vascular instability by promoting endothelial destabilisation, pericyte loss, and pathological angiogenesis, thus potentiating vascular leakage and inflammation. It also sensitises blood vessels to the activity of VEGF-A resulting in further vascular destabilisation. Ang-2 and VEGF-A synergistically increase vascular permeability and stimulate neovascularisation.
By dual inhibition of Ang-2 and VEGF-A, faricimab reduces vascular permeability and inflammation, inhibits pathological angiogenesis and restores vascular stability.
Pharmacodynamic effect.
A suppression from baseline of median ocular free Ang-2 and free VEGF-A concentrations was observed from day 7 onwards in the six Phase III studies. Systemic pharmacodynamic effects are unlikely, supported by the absence of significant changes in free VEGF and Ang-2 concentration in plasma upon faricimab treatment in clinical studies.
nAMD.
In Phase III studies in patients with nAMD (TENAYA, LUCERNE), objective, pre-specified visual and anatomic criteria, as well as the treating ophthalmologist's clinical assessment, were used to guide treatment decisions at the disease activity assessment time points (week 20 and week 24).
Reductions in mean central subfield thickness (CST) were observed from baseline through week 48 with Vabysmo, and were comparable to those observed with aflibercept. The mean CST reduction from baseline to the primary endpoint visits (averaged at weeks 40-48) was -137 micrometre and -137 micrometre for Vabysmo dosed up to every 16 weeks (Q16W) versus -129 micrometre and -131 micrometre with aflibercept, in TENAYA and LUCERNE, respectively. In year 2, the mean CST reduction from baseline to the average of weeks 104-112 was -146.5 micrometre and -150.3 micrometre for Vabysmo dosed up to Q16W versus -146.2 micrometre and -141.6 micrometre with aflibercept, in TENAYA and LUCERNE, respectively.
There was a comparable effect of Vabysmo and aflibercept on the reduction of intraretinal fluid (IRF), subretinal fluid (SRF), and pigment epithelial detachment (PED). At the primary endpoint visits (min-max, weeks 40-48), the proportion of patients in TENAYA and LUCERNE, respectively, with absence of IRF was: 76%-82% and 78%-85% in Vabysmo vs. 74%-85% and 78%-84% in aflibercept; absence of SRF: 70%-79% and 66%-78% in Vabysmo vs. 66%-78% and 62%-76% in aflibercept; absence of PED: 3%-8% and 3%-6% in Vabysmo vs. 8%-10% and 7%-9% in aflibercept. These reductions in IRF, SRF, and PED were similar at year 2 (weeks 104-108).
At week 48, there was comparable change in total CNV lesion area from baseline across treatment arms (0.0 mm2 and 0.4 mm2 in Vabysmo vs. 0.4 mm2 and 1.0 mm2 in aflibercept, in TENAYA and LUCERNE, respectively). There was a comparable reduction in CNV leakage area from baseline across treatment arms (-3.8 mm2 and -3.2 mm2 in Vabysmo and -3.0 mm2 and -2.2 mm2 in aflibercept, in TENAYA and LUCERNE, respectively).
DMO.
In Phase III studies in patients with DMO (YOSEMITE and RHINE), anatomic parameters related to macular oedema were part of the disease activity assessments guiding treatment decisions.
The reductions in mean CST from baseline were numerically greater in patients treated with Vabysmo every 8 weeks (Q8W) and Vabysmo up to Q16W adjustable dosing as compared to aflibercept Q8W from week 4 to week 100 in both YOSEMITE and RHINE. Greater proportions of patients in both Vabysmo arms achieved absence of IRF and absence of DMO (defined as reaching CST below 325 micrometre) as measured on Spectral Domain Optical Coherence Tomography (SD-OCT) over time in both studies, compared to the aflibercept arm. Comparable reductions in SRF were observed across both Vabysmo and aflibercept treatment arms over time in both studies.
The mean reduction of CST from baseline to the primary endpoint visits (averaged at weeks 48-56) was 207 micrometre and 197 micrometre in patients treated with Vabysmo Q8W and Vabysmo up to Q16W adjustable dosing as compared to 170 micrometre in aflibercept Q8W patients in YOSEMITE; results were 196 micrometre, 188 micrometre and 170 micrometre, respectively in RHINE. These mean CST reductions were maintained through year 2. The proportion of patients with absence of DMO at primary endpoint visits (min-max, weeks 48-56) were 77%-87% and 80%-82% in patients treated with Vabysmo Q8W and Vabysmo up to Q16W adjustable dosing, as compared to 64%-71% in aflibercept Q8W patients in YOSEMITE; results were 85%-90%, 83%-87%, and 71%-77%, respectively in RHINE. These results were maintained through year 2.
The proportions of patients with absence of IRF at primary endpoint visits (min-max, weeks 48-56) were 42%-48% and 34%-43% in patients treated with Vabysmo Q8W and Vabysmo up to Q16W adjustable dosing, as compared to 22%-25% in aflibercept Q8W patients in YOSEMITE; results were 39%-43%, 33%-41%, and 23%-29%, respectively in RHINE. These results were maintained through year 2.
RVO.
In Phase III studies in patients with branch retinal vein occlusion (BRVO; BALATON) and central/hemiretinal vein occlusion (C/HRVO; COMINO), reductions in mean CST were observed from baseline to week 24 with Vabysmo every 4 weeks (Q4W) and were comparable to those seen with aflibercept Q4W. The mean CST reduction from baseline to week 24 was 311.4 micrometre for Vabysmo versus 304.4 micrometre for aflibercept, and 461.6 micrometre for Vabysmo Q4W versus 448.8 micrometre for aflibercept Q4W, in BALATON and COMINO, respectively. CST reductions were maintained through week 72 when patients moved to a Vabysmo up to Q16W adjustable dosing regimen.
Comparable proportions of patients in both Vabysmo and aflibercept arms achieved absence of IRF, absence of SRF and absence of macular oedema (defined as reaching CST below 325 micrometre) over time through week 24, in both studies. These results were maintained through week 72 when patients moved to a Vabysmo up to Q16W adjustable dosing regimen.
In BALATON, at week 24, the proportion of patients with absence of macular oedema was 95.3% in patients treated with Vabysmo Q4W versus 93.9% in patients treated with aflibercept Q4W; the proportion of patients with absence of IRF was 72.5% in patients treated with Vabysmo Q4W versus 66% in patients treated with aflibercept Q4W. The proportion of patients with absence of SRF was 91.3% in patients in the Vabysmo Q4W arm, versus 90.3% in patients in the aflibercept Q4W arm.
In COMINO, at week 24, the proportion of patients with absence of macular oedema was 93.7% in patients treated with Vabysmo Q4W versus 92% in patients treated with aflibercept Q4W. The proportion of patients with absence of IRF was 76.2% in patients treated with Vabysmo Q4W versus 70.8% in patients treated with aflibercept Q4W; the proportion of patients with absence of SRF was 96.4% in patients treated with Vabysmo Q4W versus 93.4% in patients treated with aflibercept Q4W.
Clinical trials.
Treatment of nAMD.
The safety and efficacy of Vabysmo (faricimab) were assessed in two randomised, multi-centre, double-masked, active comparator-controlled studies in patients with nAMD, TENAYA (NCT03823287) and LUCERNE (NCT03823300). A total of 1,329 patients were enrolled in these studies, with 1,135 (85%) patients completing the studies through week 112. A total of 1,326 patients received at least one dose (664 with Vabysmo). Patient ages ranged from 50 to 99 with a mean of 75.9 years.
In both studies, patients were randomised in a 1:1 ratio to one of two treatment arms:
Vabysmo 6 mg up to Q16W after four initial monthly doses;
Aflibercept 2 mg Q8W after three initial monthly doses.
After the first four monthly doses (weeks 0, 4, 8, and 12) patients randomised to the Vabysmo arm received Q16W, every 12 weeks (Q12W) or Q8W dosing based on an assessment of disease activity at weeks 20 and 24, using objective pre-specified visual and anatomic criteria as well as the treating ophthalmologist's clinical assessment. Patients remained on these fixed dosing intervals until week 60 without supplemental therapy. From week 60 onwards, patients in the Vabysmo arm moved to an adjustable dosing regimen, where the dosing interval could be increased in up to 4-week increments (up to Q16W) or could be decreased by up to 8-week increments (up to Q8W) based on an automated objective assessment of pre-specified visual and anatomic disease activity criteria. Patients in the aflibercept arm remained on Q8W dosing throughout the study period. Both studies were 112 weeks in duration.
The primary efficacy endpoint was the change from baseline in BCVA based on an average at weeks 40, 44, and 48, measured by the Early Treatment Diabetic Retinopathy Study (ETDRS) Letter Score. In both studies, Vabysmo up to Q16W treated patients had a comparable mean change from baseline in BCVA, as the patients treated with aflibercept Q8W at year 1.
Meaningful vision gains from baseline were seen through week 112 in both treatment arms. Detailed results of both studies are shown in Table 2, Table 3, Figure 1, and Figure 2.
The proportion of patients on each of the different treatment intervals at week 48 in TENAYA and LUCERNE, respectively was: Q16W: 46%, 45%; Q12W: 34%, 33%; Q8W: 20%, 22%.
The proportion of patients on each of the different treatment intervals at week 112 in TENAYA and LUCERNE, respectively was: Q16W: 59%, 67%; Q12W: 15%, 14%; Q8W: 26%, 19%.
See Table 2, Table 3, Figure 1 and Figure 2.


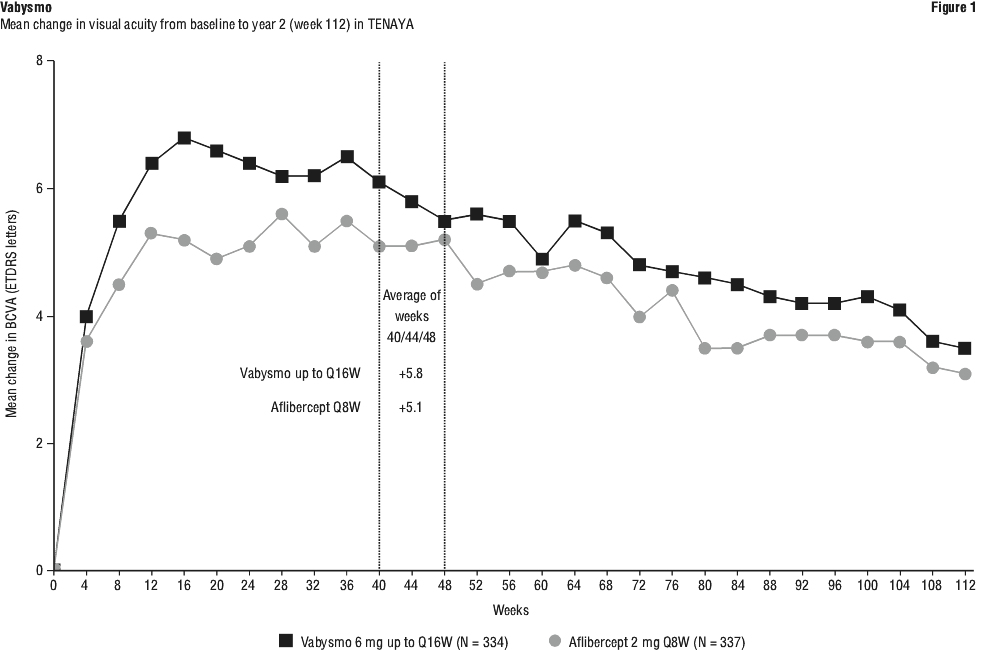
 In both TENAYA and LUCERNE, improvements from baseline in BCVA and CST at week 60 were comparable across the two treatment arms and consistent with those seen at week 48.
In both TENAYA and LUCERNE, improvements from baseline in BCVA and CST at week 60 were comparable across the two treatment arms and consistent with those seen at week 48.
Efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, lesion type, lesion size) in each study, and in the pooled analysis, were consistent with the results in the overall populations.
In both studies, Vabysmo up to Q16W demonstrated clinically meaningful improvements from baseline to week 48 in the National Eye Institute Visual Function Questionnaire (NEI VFQ-25) composite score that was comparable to aflibercept Q8W. Patients in Vabysmo arms in TENAYA and LUCERNE achieved a ≥ 4 point improvement from baseline in the NEI VFQ-25 composite score at week 48. At week 112, the mean improvement from baseline for patients receiving Vabysmo was 1.36 in TENAYA and 3.78 in LUCERNE.
Treatment of DMO.
The safety and efficacy of Vabysmo were assessed in two randomised, multi-centre, double-masked, active comparator-controlled 2-year studies (YOSEMITE and RHINE) in patients with DMO. A total of 1,891 patients were enrolled in the two studies with 1622 (85.8%) patients completing the studies through week 100. A total of 1,887 were treated with at least one dose through week 56 (1,262 with Vabysmo). Patient ages ranged from 24 to 91 with a mean of 62.2 years. The overall population included both anti-VEGF naive patients (78%) and patients who had been previously treated with a VEGF inhibitor prior to study participation (22%). In both studies, patients were randomised in a 1:1:1 ratio to one of the three treatment regimens:
Vabysmo 6 mg Q8W after the first 6 monthly doses.
Vabysmo 6 mg up to Q16W adjustable dosing administered in 4, 8, 12 or 16 week intervals after the first 4 monthly doses.
Aflibercept 2 mg Q8W after the first 5 monthly doses.
In the Q16W adjustable dosing arm, the dosing followed a standardised treat-and-extend approach. The interval could be increased in 4-week increments or decreased in 4- or 8-week increments based on anatomic and/or visual outcomes, using data obtained only at study drug dosing visits.
Both studies demonstrated efficacy in the primary endpoint, defined as the change from baseline in BCVA at year 1 (average of the week 48, 52, and 56 visits) as measured by the ETDRS Letter Score. In both studies, Vabysmo up to Q16W treated patients had a comparable mean change from baseline in BCVA, as the patients treated with aflibercept Q8W at year 1, and these vision gains were maintained through year 2. Detailed results of both studies are shown in Table 3, Table 4, Figure 3, and Figure 4.
After 4 initial monthly doses, the patients in the Vabysmo up to Q16W adjustable dosing arm could have received between the minimum of 6 and the maximum of 21 total injections through week 96. At week 52, 74% and 71% of patients in the Vabysmo up to Q16W adjustable dosing arm achieved a Q16W or Q12W dosing interval in YOSEMITE and RHINE, respectively (53% and 51% on Q16W, 21% and 20% on Q12W). Of these patients, 75% and 84% maintained ≥ Q12W dosing without an interval reduction below Q12W through week 96; of the patients on Q16W at week 52, 70% and 82% of patients maintained Q16W dosing without an interval reduction through week 96 in YOSEMITE and RHINE, respectively. At week 96, 78% of patients in the Vabysmo up to Q16W adjustable dosing arm achieved a Q16W or Q12W dosing interval in both studies (60% and 65% on Q16W, 18% and 14% on Q12W). 4% and 6% of patients were extended to Q8W and stayed on ≤ Q8W dosing intervals through week 96; 3% and 5% received only Q4W dosing in YOSEMITE and RHINE through to week 96, respectively.
Detailed results from the analyses of YOSEMITE and RHINE studies are listed in Table 4, Table 5 and Figures 3 and 4.



 Efficacy results in patients who were anti-VEGF treatment naive prior to study participation and in all the other evaluable subgroups (e.g. by age, gender, race, baseline HbA1c, baseline visual acuity) in each study were consistent with the results in the overall populations.
Efficacy results in patients who were anti-VEGF treatment naive prior to study participation and in all the other evaluable subgroups (e.g. by age, gender, race, baseline HbA1c, baseline visual acuity) in each study were consistent with the results in the overall populations.
Across studies, Vabysmo Q8W and up to Q16W adjustable dosing showed clinically meaningful improvements in the pre-specified efficacy endpoint of mean change from baseline to week 52 in the NEI VFQ-25 composite score that were comparable to aflibercept Q8W and exceeded the threshold of 4 points. Vabysmo Q8W and up to Q16W adjustable dosing also demonstrated clinically meaningful improvements in the pre-specified efficacy endpoint of change from baseline to week 52 in the NEI VFQ-25 near activities, distance activities, and driving scores, that were comparable to aflibercept Q8W. The magnitude of these changes corresponds to a 15-letter gain in BCVA. Comparable proportions of patients treated with Vabysmo Q8W, Vabysmo up to Q16W adjustable dosing, and aflibercept Q8W experienced a clinically meaningful improvement of ≥ 4 points from baseline to week 52 in the NEI VFQ-25 composite score, a pre-specified efficacy endpoint. These results were maintained at week 100.
An additional key efficacy outcome in DMO studies was the change in the Early Treatment Diabetic Retinopathy Study Diabetic Retinopathy Severity Scale (ETDRS-DRSS) from baseline to week 52. Of the 1,891 patients enrolled in Studies YOSEMITE and RHINE, 708 and 720 patients were evaluable for DR endpoints, respectively.
The ETDRS-DRSS scores ranged from 10 to 71 at baseline.
The majority of patients, approximately 60%, had moderate to severe non-proliferative DR (DRSS 43/47/53) at baseline.
At week 52, the proportion of patients improving by ≥ 2 steps on the ETDRS-DRSS was 43% to 46% across the Vabysmo Q8W and Vabysmo adjustable up to Q16W arms in both studies, compared to 36% and 47% in aflibercept Q8W arms of YOSEMITE and RHINE, respectively. The results at week 96 were 43% to 54% across the Vabysmo Q8W and Vabysmo adjustable up to Q16W arms in both studies, compared to 42% and 44% in aflibercept Q8W arms of YOSEMITE and RHINE, respectively.
Comparable results across the treatment arms were observed in both studies in the proportions of patients improving by ≥ 3 steps on the ETDRS-DRSS from baseline at week 52, and these results were maintained at week 96.
The results from the ≥ 2-step and ≥ 3-step ETDRS-DRSS improvement analyses from baseline at week 52 and at week 96 are shown in Tables 6 and 7.

 Among patients who had no proliferative DR at baseline (Vabysmo Q8W n = 406, Vabysmo up to Q16W adjustable dosing n = 422 and aflibercept Q8W n = 400, in pooled YOSEMITE and RHINE), the proportions of patients who developed new proliferative DR diagnosis (defined by ETDRS-DRSS 61 or worse) from baseline to week 96 were comparable between the Vabysmo Q8W, Vabysmo up to Q16W adjustable dosing and aflibercept Q8W dosed patients in pooled YOSEMITE and RHINE studies. In a descriptive analysis in the ITT population, almost no patients required vitrectomy (0 to 4 per group) or Pan-retinal Photocoagulation (PRP) (1 to 2 per group) during the two year duration of the studies.
Among patients who had no proliferative DR at baseline (Vabysmo Q8W n = 406, Vabysmo up to Q16W adjustable dosing n = 422 and aflibercept Q8W n = 400, in pooled YOSEMITE and RHINE), the proportions of patients who developed new proliferative DR diagnosis (defined by ETDRS-DRSS 61 or worse) from baseline to week 96 were comparable between the Vabysmo Q8W, Vabysmo up to Q16W adjustable dosing and aflibercept Q8W dosed patients in pooled YOSEMITE and RHINE studies. In a descriptive analysis in the ITT population, almost no patients required vitrectomy (0 to 4 per group) or Pan-retinal Photocoagulation (PRP) (1 to 2 per group) during the two year duration of the studies.
DR treatment effects in the subgroup of patients who were anti-VEGF naive prior to study participation were comparable to those observed in the overall DR evaluable population. Treatment effects in evaluable subgroups (e.g. by age, gender, race, baseline HbA1c, and baseline visual acuity) in each study were generally consistent with the results in the overall population.
Treatment effects in subgroups by DR severity at baseline were different and showed the greatest ≥ 2-step DRSS improvements among patients with moderately severe and severe non-proliferative DR (Vabysmo Q8W n = 188, Vabysmo up to Q16 adjustable dosing n = 190 and aflibercept Q8W n = 170, in pooled YOSEMITE and RHINE) with approximately 90% of patients achieving improvements. These results were comparable across the study arms, and comparable in overall and anti-VEGF treatment-naive populations.
Treatment of macular oedema secondary to RVO.
The safety and efficacy of Vabysmo were assessed in two randomised, multi-centre, double-masked, 72-week long studies in patients with macular oedema secondary to BRVO (BALATON) or C/HRVO (COMINO). Active comparator-controlled data are available through month 6.
A total of 1,282 patients (553 in BALATON and 729 in COMINO) were enrolled in the two studies, with 1,276 patients treated with at least one dose through week 24 (641 with Vabysmo).
In both studies, patients were randomised in a 1:1 ratio to one of two treatment arms:
Vabysmo 6 mg Q4W for six consecutive monthly doses.
Aflibercept 2 mg Q4W for six consecutive monthly doses.
After six initial monthly doses, patients initially randomised to the 2 mg aflibercept arm moved to the 6 mg Vabysmo arm and could receive 6 mg Vabysmo up to Q16W adjustable dosing regimen, where the dosing interval could be increased in 4-week increments up to Q16W or decreased by 4-, 8- or 12-weeks based on an automated objective assessment of pre-specified visual and anatomic disease activity criteria.
Both studies assessed and demonstrated efficacy using a primary endpoint of change from baseline in BCVA at week 24, measured by the ETDRS Letter Score. In both studies, Vabysmo Q4W treated patients had a non-inferior mean change from baseline in BCVA at week 24, compared to patients treated with aflibercept Q4W. These vision gains were maintained through week 72 when patients moved to a Vabysmo up to Q16W adjustable dosing regimen.
Between week 24 and week 68, 81.5% and 74.0% of the patients receiving Vabysmo 6 mg up to Q16W adjustable dosing regimen achieved a Q16W or Q12W dosing interval in BALATON and COMINO, respectively. Of these patients, 72.1% and 61.6% completed at least one cycle of Q12W, and maintained Q16W or Q12W dosing interval without an interval reduction below Q12W through week 68 in BALATON and COMINO, respectively; 1.2% and 2.5% of the patients received only Q4W dosing through week 68 in BALATON and COMINO, respectively.
Detailed results of both studies are shown in Table 8, Table 9, Figure 5 and Figure 6.


 Vabysmo 6 mg up to Q16W adjustable dosing started at week 24 but not all patients received Vabysmo at week 24.
Vabysmo 6 mg up to Q16W adjustable dosing started at week 24 but not all patients received Vabysmo at week 24.
 Vabysmo 6 mg up to Q16W adjustable dosing started at week 24 but not all patients received Vabysmo at week 24.
Vabysmo 6 mg up to Q16W adjustable dosing started at week 24 but not all patients received Vabysmo at week 24.
Immunogenicity.
Immunogenicity assay results are highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of incidence of antibodies to Vabysmo with the incidence of antibodies to other products may be misleading.
5.2 Pharmacokinetic Properties
Absorption.
Vabysmo is administered intravitreally (IVT) to exert local effects in the eye. There have been no clinical studies performed with other routes of administration.
Based on a population pharmacokinetic analysis (including nAMD and DMO N = 2,246), maximum free (unbound to VEGF-A and Ang-2) faricimab plasma concentrations (Cmax) are estimated to occur approximately 2 days post-dose. Mean (± SD) plasma Cmax are estimated 0.23 (0.07) microgram/mL and 0.22 (0.07) microgram/mL respectively in nAMD and in DMO/DR patients. After repeated administrations, mean plasma free faricimab trough concentrations are predicted to be 0.002-0.003 microgram/mL for Q8W dosing.
Faricimab exhibited dose-proportional pharmacokinetics (based on Cmax and AUC) over the dose range 0.5 mg-6 mg. No accumulation of faricimab was apparent in the vitreous or in plasma following monthly dosing.
Pharmacokinetic analysis of patients with nAMD, DMO, and RVO (n = 2,977) has shown that the pharmacokinetics of faricimab are comparable in nAMD, DMO and RVO patients.
Distribution.
Maximum plasma free faricimab concentrations are predicted to be approximately 600 and 6000-fold lower than in aqueous and vitreous humour respectively and are below the binding affinity for VEGF-A and Ang-2. Therefore, systemic pharmacodynamic effects are unlikely, further supported by the absence of significant changes in free VEGF and Ang-2 concentration in plasma upon faricimab treatment in clinical studies.
Population pharmacokinetic analysis has shown an effect of age and body weight on ocular or systemic pharmacokinetics of faricimab respectively. Both effects were considered not clinically meaningful; no dose adjustment is needed.
In the clinical studies with faricimab, no significant suppression in free VEGF and Ang-2 was observed in plasma.
Metabolism.
The metabolism of faricimab has not been directly studied, as monoclonal antibodies are cleared principally by catabolism.
Elimination.
The faricimab plasma concentration-time profile declined in parallel with the vitreous and aqueous concentration-time profiles. The estimated mean ocular half-life and apparent systemic half-life of faricimab is approximately 7.5 days after IVT administration.
Special populations.
Renal impairment.
No formal pharmacokinetic study has been conducted in patients with renal impairment. Pharmacokinetic analysis of patients in all clinical studies of which 63% had renal impairment (mild 38%, moderate 23%, and severe 2%), revealed no differences with respect to systemic pharmacokinetics of faricimab after intravitreal administration of Vabysmo.
Hepatic impairment.
No formal pharmacokinetic study has been conducted in patients with hepatic impairment. However, no special considerations are needed in this population because metabolism occurs via proteolysis and does not depend on hepatic function.
Use in the elderly.
In the six Phase III clinical studies, approximately 58% (1,496/2,571) of patients randomised to treatment with Vabysmo were ≥ 65 years of age. Population pharmacokinetic analysis has shown an effect of age on ocular pharmacokinetics of faricimab, which was not considered clinically meaningful.
Paediatric use.
The safety and efficacy of Vabysmo in paediatric patients have not been established.
5.3 Preclinical Safety Data
Genotoxicity.
No studies have been performed to establish the mutagenic potential of faricimab.
Carcinogenicity.
No carcinogenicity studies have been performed to establish the carcinogenic potential of faricimab.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, acetic acid 30% (for pH adjustment), methionine, polysorbate 20, sodium chloride, sucrose, water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicine must not be mixed with other medicines.
6.3 Shelf Life
Vabysmo vial.
30 months.
Vabysmo pre-filled syringe.
24 months.
6.4 Special Precautions for Storage
Store at 2°C to 8°C.
Keep the vial in the outer carton in order to protect from light.
Keep the pre-filled syringe in the sealed tray in the original carton in order to protect from light.
Do not freeze. Do not shake.
Prior to use, the unopened vial or pre-filled syringe of Vabysmo may be kept at 20°C to 25°C in the original carton for up to 24 hours.
Ensure that the injection is given immediately after preparation of the dose.
6.5 Nature and Contents of Container
Vabysmo vial.
0.24 mL sterile, preservative-free solution in a glass vial with a coated rubber stopper sealed with an aluminum cap with a yellow plastic flip-off disk.
Pack size of 1 vial and 1 blunt transfer filter needle (18-gauge x 1½ inch, 1.2 mm x 40 mm).
Vabysmo pre-filled syringe.
Solution for injection in a pre-filled syringe consisting of a glass barrel with a dose mark, a rubber stopper, and a tamper-evident closure cap (including a rigid tip cap, a rubber tip cap and a luer lock 22 adapter). The container is assembled with a plunger rod and an extended finger flange. Each pre-filled syringe contains 21 mg faricimab in 0.175 mL solution.
Pack size of 1 pre-filled syringe and 1 injection filter needle (30-gauge x ½ inch, 0.30 mm x 12.7 mm, extra thin wall).
6.6 Special Precautions for Disposal
Preparation for administration.
Vabysmo is a sterile, preservative-free, clear to opalescent, colourless to brownish-yellow solution.
Do not shake.
Vabysmo vial.
The vial contains more than the recommended dose of 6 mg. The fill volume of the vial (0.24 mL) is not to be used in total. The excess volume should be expelled prior to injection. Injecting the entire volume of the vial results in overdose. The injection dose must be set to the 0.05 mL dose mark, i.e. 6 mg of Vabysmo.
The contents of the vial and transfer filter needle are sterile and for single use in one patient only. Do not use if the packaging, vial and/or transfer filter needle are damaged or expired.
Vabysmo pre-filled syringe.
The pre-filled syringe is for single use in one eye only. Open the sterile pre-filled syringe under aseptic conditions only. The solution should be inspected visually prior to administration. If particulates or cloudiness are visible, the pre-filled syringe must not be used.
The pre-filled syringe contains more than the recommended dose of 6 mg faricimab (equivalent to 0.05 mL). Each pre-filled syringe contains 21 mg faricimab in 0.175 mL solution. The excess volume must be expelled prior to injection.
Do not use if the packaging, pre-filled syringe and/or injection filter needle are damaged or expired.
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.6.7 Physicochemical Properties
Chemical structure.
Faricimab is a humanised bispecific antibody produced in mammalian Chinese Hamster Ovary (CHO) cell culture by recombinant DNA technology. It consists of two different heavy chains (VEGF-HC: HC1 with 452 amino acid residues, Ang-2-HC: HC2 with 462 amino acid residues; without C-terminal lysine) and two different light chains (VEGF-LC: LC1 with 214 amino acid residues, Ang-2-LC: LC2 with 213 amino acid residues) with inter- and intra-chain disulfide bonds that are typical for IgG1 antibodies plus an additional disulfide bridge in the CH3-CH3 interface. The approximate molecular weight of intact faricimab is 146 kDa.
CAS number.
1607793-29-2.7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine.
Summary Table of Changes
