Notes
Distributed by Southern XP Pty Ltd
1 Name of Medicine
Sodium valproate.
2 Qualitative and Quantitative Composition
Valproate SXP solution for injection/infusion is supplied in 300 mg/3 mL or 400 mg/4 mL vials.
Each 300 mg/3 mL vial of Valproate SXP contains 300 mg sodium valproate in 3 mL solution.
Each 400 mg/4 mL vial of Valproate SXP contains 400 mg sodium valproate in 4 mL solution.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Valproate SXP is supplied as a sterile, clear and colourless solution for intravenous injection/infusion in a single dose vial. The product is available in 300 mg/3 mL and 400 mg/4 mL strengths.
4.1 Therapeutic Indications
Valproate SXP is used for the treatment of patients with epilepsy or mania, who would normally be maintained on oral sodium valproate, and for whom oral therapy is temporarily not possible.
4.2 Dose and Method of Administration
Sodium valproate tablets, liquid and syrup are unavailable in the Valproate SXP brand.
Valproate SXP may be given by direct slow intravenous injection or by slow intravenous infusion in 0.9% NaCl (normal saline), 5% glucose solution or glucose saline, using a separate intravenous line. The recommended concentration of the intravenous infusion solution is 4 mg/mL, with 8 mg/mL being the maximum concentration.
Valproate SXP should not be administered at the same time as other intravenous additives via the same IV line. The intravenous solution is suitable for infusion by PVC, polyethylene or glass containers. Valproate SXP should be replaced by oral sodium valproate therapy as soon as practicable.
Each vial of Valproate SXP is for single dose injection only. To reduce microbiological hazard, use as soon as practicable after dilution. If storage is necessary, hold at 2 to 8 °C for not more than 24 hours. Valproate SXP is intended for use in one patient on one occasion only, any unused portion should be discarded. Never administer Valproate SXP other than by the intravenous route (see Section 4.4 Special Warnings and Precautions for Use).
Monotherapy.
Daily dosage requirements vary according to age and body weight.
Adults.
Patients already satisfactorily treated with Valproate SXP may be continued at their current dosage using continuous infusion. For example, a patient stabilised on 25 mg/kg administered daily should be continued with an infusion at the rate of 1 mg/kg/hr.
Other patients may be given a slow intravenous injection over 3-5 minutes, usually 400-800 mg depending on body weight (up to 10 mg/kg) followed by continuous infusion of 1-2 mg/kg/hr up to a maximum of 2500 mg/day, according to the patient's clinical response.
Use in children.
The daily requirement for children is usually in the range 20-30 mg/kg/day and method of administration is as above. Where adequate control is not achieved within this range the dose may be increased up to 40 mg/kg/day but only in patients in whom plasma valproic acid levels can be monitored. Above 40 mg/kg/day clinical chemistry and haematological parameters should be monitored.
Combined therapy.
When starting Valproate SXP in patients already on other anticonvulsants, these should be tapered slowly: initiation of Valproate SXP therapy should be gradual, with the target dose being reached after about 2 weeks. In certain cases it may be necessary to raise the dose by 5 to 10 mg/kg/day when used in combination with anticonvulsants which induce liver enzyme activity, e.g. phenytoin, phenobarbital (phenobarbitone) and carbamazepine. Once known enzyme inducers have been withdrawn it may be possible to maintain symptom control on a reduced dose of Valproate SXP. When barbiturates are being administered concomitantly the dosage of barbiturate should be reduced if sedation is observed (particularly in children). (See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.)
Use in hepatic impairment.
Liver dysfunction, including hepatic failure resulting in fatalities, has occurred in patients whose treatment included valproic acid or sodium valproate (see Section 4.4 Special Warnings and Precautions for Use).
Use in renal impairment.
Lower doses may be required since free drug levels may be high owing to lowered serum albumin and poor urinary excretion of free drug metabolites (see Section 4.4 Special Warnings and Precautions for Use). Dosage should be adjusted according to clinical monitoring since monitoring of plasma concentrations may be misleading.
Use in the elderly.
Although the pharmacokinetics of sodium valproate are modified in the elderly, they have limited clinical significance and dosage should be determined by control of symptoms. The volume of distribution is increased in the elderly and because of decreased binding to serum albumin, the proportion of free drug is increased. This will affect the clinical interpretation of plasma valproic acid levels.
Use in female children, women of child bearing potential and pregnant women.
See Section 4.4 Special Warnings and Precautions for Use.
Sodium valproate must be initiated and supervised by a specialist experienced in the management of epilepsy or bipolar disorder. Valproate should not be used in female children and women of childbearing potential unless other treatments are ineffective or not tolerated (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use; Section 4.6 Fertility, Pregnancy and Lactation).
In the exceptional circumstances when valproate is the only treatment option during pregnancy in epileptic women, valproate should preferably be prescribed as monotherapy, at the lowest effective dose.
The daily dose of non-prolonged release formulations should be divided into at least two single doses during pregnancy (see Section 4.6 Fertility, Pregnancy and Lactation).
Estrogen containing products.
The enzyme inducing effect of valproate is appreciably less than that of certain other anticonvulsants and loss of efficacy of oral contraceptive agents does not appear to be a problem.
However, estrogen containing products, including estrogen containing hormonal contraceptives, may increase the clearance of valproate, which may result in decreased serum concentration of valproate and potentially decreased valproate efficacy. Prescribers should monitor clinical response (seizure control or mood control) when initiating or discontinuing estrogen-containing products. Consider monitoring of valproate serum levels. (See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.)
General considerations.
Optimum dosage is mainly determined by control of symptoms and routine measurement of plasma levels is unnecessary. However, a method for measurement of plasma levels is available and may be helpful where there is poor control or side effects are suspected.4.3 Contraindications
Valproate SXP is contraindicated in the following situations:
Treatment of epilepsy.
In pregnancy unless there is no suitable alternative treatment (see Section 4.4 Special Warnings and Precautions for Use; Section 4.6 Fertility, Pregnancy and Lactation).
In women of childbearing potential, unless the physician has provided information in regards to the potential effects of valproate during pregnancy and recommendations on the use of valproate.
Treatment of mania.
In pregnancy (see Section 4.4 Special Warnings and Precautions for Use; Section 4.6 Fertility, Pregnancy and Lactation).
In women of childbearing potential, unless the physician has provided information in regards to the potential effects of valproate during pregnancy and the recommendations on the use of valproate.
All indications.
Pre-existing, acute or chronic hepatic dysfunction or family history of severe hepatitis, particularly medicine related. Known hypersensitivity to the medicine. Known urea cycle disorders (see Section 4.4 Special Warnings and Precautions for Use). Known systemic primary carnitine deficiency with uncorrected hypocarnitinemia (see Section 4.4 Special Warnings and Precautions for Use). Known hepatic porphyria. Patients known to have mitochondrial disorders caused by mutations in the nuclear gene encoding mitochondrial enzyme polymerase γ (POLG e.g. Alpers-Huttenlocher syndrome) and in children under two years of age who are suspected of having POLG-related disorder.
Valproate SXP should not be injected intramuscularly as it may produce tissue necrosis.4.4 Special Warnings and Precautions for Use
Pregnancy and women of childbearing potential.
Valproate has a high teratogenic potential and children exposed in utero to valproate have a high risk for congenital malformations and neurodevelopmental disorders (see Section 4.6 Fertility, Pregnancy and Lactation).
Valproate should not be used in female children and women of childbearing potential unless other treatments are ineffective or not tolerated (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use; Section 4.6 Fertility, Pregnancy and Lactation).
In the exceptional circumstance when valproate is the only treatment option available for women of childbearing potential, the physician must ensure that:
Individual circumstances are evaluated and discussed with the patient. This is to guarantee the patient's engagement and understanding of the therapeutic options together with the risks and the measures needed to mitigate the risks.
The potential for pregnancy is assessed for all female patients.
The patient understands the risks of congenital malformations and neurodevelopmental disorders including the magnitude of these risks for children exposed to valproate in utero.
The patient understands the risk of lower weight at birth for the gestational age for children exposed to valproate in utero (see Section 4.6 Fertility, Pregnancy and Lactation).
The patient understands the need to undergo pregnancy testing prior to initiation of treatment and during treatment, as needed.
The patient is counselled regarding contraception, and that the patient is capable of complying with the need to use effective contraception (see Section 4.4 Special Warnings and Precautions for Use, Pregnancy and women of childbearing potential, Contraception), without interruption during the entire duration of treatment with valproate.
The patient understands the need for regular (at least annual) review of treatment by a specialist experienced in the management of epilepsy, or bipolar disorders.
The patient understands the need to consult her physician as soon as she is planning pregnancy to ensure timely discussion and switching to alternative treatment options prior to conception, and before contraception is discontinued.
The patient understands the need to urgently consult her physician in case of pregnancy.
The patient understands the hazards and necessary precautions associated with valproate use.
These conditions also concern women who are not currently sexually active unless the prescriber considers that there are compelling reasons to indicate that there is no risk of pregnancy.
Pharmacist or other healthcare professional must:
Review the CMI with the patient (or parent/caregiver).
Advise the patient (or parent/caregiver) not to stop valproate medication and to immediately contact a specialist in case of planned or suspected pregnancy.
Female children.
The prescribers must ensure that parents/caregivers of female children understand the need to contact the specialist once the female child using valproate experiences menarche.
The prescriber must ensure that parents/caregivers of female children who have experienced menarche are provided with comprehensive information about the risks of congenital malformations and neurodevelopmental disorders including the magnitude of these risks for children exposed to valproate in utero. The prescriber must also inform them about the risk of lower weight at birth for the gestational age.
In patients who experience menarche, the prescribing specialist must reassess the need for valproate therapy annually and consider alternative treatment options. If valproate is the only suitable treatment, the need for using effective contraception program should be discussed. Every effort should be made by the specialist to switch the female children to alternative treatment before they reach adulthood.
Pregnancy test.
Pregnancy must be excluded before the start of treatment with valproate. Treatment with valproate must not be initiated in women of child bearing potential without a negative pregnancy test (plasma pregnancy test) result, confirmed by a health care provider, to rule out unintended use in pregnancy.
Contraception.
Women of childbearing potential who are prescribed valproate must use effective contraception, without interruption during the entire duration of treatment with valproate. These patients must be provided with comprehensive information on pregnancy prevention. If they are not using effective contraception they should be referred for contraceptive advice.
At least one effective method of contraception (preferably a user-independent form such as an intrauterine device or implant) or two complementary forms of contraception including a barrier method should be used.
Individual circumstances should be evaluated in consultation with the patient when choosing contraception method. This will increase patient engagement and compliance with the chosen contraceptive measures.
Even if patient has amenorrhea she must follow all the advice on effective contraception.
Annual treatment review.
The specialist should review, at least annually, whether valproate is the most suitable treatment for the patient.
Pregnancy planning.
For the epilepsy indication, if a patient on valproate is planning to become pregnant, a specialist experienced in the management of epilepsy must reassess the therapy and consider alternative treatment options. Every effort should be made to switch to appropriate alternative treatment prior to conception, and before contraception is discontinued (see Section 4.6 Fertility, Pregnancy and Lactation). If switching is not possible, the patient should receive further counselling regarding the risks of valproate for the unborn child to support her informed decision making regarding family planning.
For the bipolar disorder indication, if a woman is planning to become pregnant, a specialist experienced in the management of bipolar disorder must be consulted. Treatment with valproate should be discontinued prior to conception, and before contraception is discontinued. If needed, alternative treatment options should be considered.
In case of pregnancy.
If a woman using valproate becomes pregnant, she must be immediately referred to a specialist to reevaluate treatment with valproate and consider alternative options. The patients with a valproate exposed pregnancy and their partners should be referred to a specialist experienced in pre-natal medicine for evaluation and counselling regarding the exposed pregnancy (see Section 4.6 Fertility, Pregnancy and Lactation).
Use in males of reproductive potential.
A retrospective observational study indicates an increased risk of neurodevelopmental disorders (NDDs) in children born to men treated with valproate in the 3 months prior to conception, compared to those treated with lamotrigine or levetiracetam (see Section 4.6 Fertility, Pregnancy and Lactation).
Despite study limitations, by way of precaution, the prescriber should inform the male patients of this potential risk. The prescriber should discuss with the patient the need for effective contraception, including for the female partner, while using valproate and for 3 months after stopping the treatment. The risk to children born to men stopping valproate at least 3 months prior to conception (i.e. allowing a new spermatogenesis without valproate exposure) is not known.
The male patient should be advised:
not to donate sperm during treatment and for 3 months after stopping the treatment;
of the need to consult his doctor to discuss alternative treatment options, as soon as he is planning to father a child, and before discontinuing contraception;
that he and his female partner should contact their doctor for counselling in case of pregnancy if he used valproate within 3 months prior to conception.
The male patient should also be informed about the need for regular (at least annual) review of treatment by a specialist experienced in the management of epilepsy or bipolar disorder. The specialist should at least annually review whether valproate is the most suitable treatment for the patient. During this review, the specialist should ensure the male patient has acknowledged the risk and understood the precautions needed with valproate use.
Hepatic dysfunction.
Conditions of occurrence.
Severe liver damage and/or hepatic failure resulting in fatalities have occurred in patients whose treatment included valproic acid or sodium valproate. Patients most at risk of liver injury are those on multiple anticonvulsant therapy including those on cannabidiol (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 4.8 Adverse Effects (Undesirable Effects)). Dose adjustment or discontinuation of valproate or the concomitant drug should be considered if transaminase elevations occur.
Children, particularly those under the age of 3 years and those with congenital metabolic disorders including mitochondrial disorders such as carnitine deficiency, urea cycle disorders, POLG mutations (see Section 4.4 Special Warnings and Precautions for Use) or degenerative disorders, organic brain disease or severe seizure disorders associated with brain damage and/or mental retardation are also at higher risk of liver injury.
The incidents usually occurred during the first six months of therapy, the period of maximum risk being 2 to 12 weeks, and usually involved multiple anticonvulsant therapy. Monotherapy is to be preferred in this group of patients.
Suggestive signs.
Clinical symptoms are usually more helpful than laboratory investigations in the early stages of hepatic failure. Jaundice, serious or fatal hepatotoxicity may be preceded by nonspecific symptoms, usually of sudden onset, such as loss of seizure control, malaise, asthenia, weakness, lethargy, facial oedema, anorexia, vomiting, abdominal pain, drowsiness, jaundice. In patients with epilepsy, recurrence of seizures can occur. These are an indication for immediate withdrawal of the medicine. Patients should be monitored closely for the appearance of these symptoms. Patients (and their family and carers) should be instructed to immediately report any such signs to the clinician for investigation should they occur. Investigations including clinical examination and laboratory assessment of liver functions should be undertaken immediately.
Detection.
Although published evidence does not establish which, if any investigation could predict this possible adverse effect, liver function tests should be performed (especially in patients at risk) prior to therapy and frequently thereafter until 6 months after the controlling dose is reached, when less frequent monitoring may be appropriate. It is also advisable to monitor tests which reflect protein synthesis, e.g. prothrombin time, serum fibrinogen and albumin levels, especially in those who seem most at risk and those with a prior history of hepatic disease.
As with most antiepileptic drugs, a slight increase in liver enzymes may be noted, particularly at the beginning of therapy. They are transient and isolated. More extensive biological investigations (including prothrombin rate) are recommended in those patients. An adjustment of dosage may be considered when appropriate and tests should be repeated as necessary.
Raised liver enzymes are not uncommon during treatment with sodium valproate, particularly if used in conjunction with other anticonvulsants, and are usually transient or respond to dosage reduction. Patients with such biochemical abnormalities should be reassessed clinically and tests of liver function should be monitored more frequently. An abnormally low prothrombin rate, particularly in association with other relevant abnormalities (significant decrease in fibrinogen and coagulation factors; increased bilirubin level and raised transaminases) requires cessation of treatment and the substitution of alternative medicines to avoid precipitating convulsions. Uneventful recovery has been recorded in several cases where therapy with sodium valproate has ceased, but death has occurred in some patients in spite of the medicine being withdrawn. Any concomitant use of salicylates should be stopped, since they employ the same metabolic pathway.
Patients with known or suspected mitochondrial disease.
Valproate may trigger or worsen clinical signs of underlying mitochondrial diseases caused by mutations of mitochondrial DNA as well as the nuclear-encoded POLG gene. In particular, acute liver failure and liver-related deaths have been associated with valproate treatment at a higher rate in patients with hereditary neurometabolic syndromes caused by mutations in the gene for mitochondrial enzyme polymerase γ (POLG e.g. Alpers-Huttenlocher syndrome).
POLG-related disorders should be suspected in patients with a family history or suggestive symptoms of a POLG-related disorder, including but not limited to un-explained encephalopathy, refractory epilepsy (focal, myoclonic), status epilepticus at presentation, developmental delays, psychomotor regression, axonal sensorimotor neuropathy, myopathy cerebellar ataxia, ophthalmoplegia, or complicated migraine with occipital aura. POLG mutation testing should be performed in accordance with current clinical practice for the diagnostic evaluation of such disorders.
Urea cycle disorders and risk of hyperammonemia.
When an urea cycle enzymatic deficiency is suspected, metabolic investigations should be performed prior to treatment because of the risk of hyperammonemia with valproate (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
Patients at risk of hypocarnitinemia.
Valproate administration may trigger occurrence or worsening of hypocarnitinemia that can result in hyperammonaemia (that may lead to hyperammonemic encephalopathy). Other symptoms such as liver toxicity, hypoketotic hypoglycaemia, myopathy including cardiomyopathy, rhabdomyolysis, Fanconi syndrome have been observed, mainly in patients with risk factors for hypocarnitinemia or pre-existing hypocarnitinemia. Valproate may decrease carnitine blood and tissue levels and therefore impair mitochondrial metabolism including the mitochondrial urea cycle. Patients at increased risk for symptomatic hypocarnitinemia when treated with valproate include patients with metabolic disorders including mitochondrial disorders related to carnitine, impairment in carnitine nutritional intake, patients younger than 10 years old, concomitant use of pivalate-conjugated medicines or other antiepileptics.
Patients should be warned to report immediately any signs of hyperammonemia such as ataxia, impaired consciousness, vomiting for further investigation. Carnitine supplementation should be considered when symptoms of hypocarnitinemia are observed.
Patients with known systemic primary carnitine deficiency and corrected for hypocarnitinemia should be treated with valproate only if the benefits of valproate treatment outweigh the risks in these patients and there is no suitable therapeutic alternative. In these patients, close monitoring for recurrence of hypocarnitinemia should be implemented.
Patients with an underlying carnitine palmitoyltransferase (CPT) type II deficiency should be warned of the greater risk of rhabdomyolysis when taking valproate. Carnitine supplementation should be considered in these patients.
Pancreatitis.
Cases of life-threatening pancreatitis have been reported in both children and adults receiving sodium valproate. Some cases have occurred shortly after initial use while others have occurred after several years of use. There have also been cases in which pancreatitis recurred after rechallenge with sodium valproate. Some of the cases have been described as haemorrhagic with a rapid progression from initial symptoms to death. In clinical trials, there were two cases of pancreatitis without alternative aetiology in 2416 patients, representing 1044 patient-years experience. Young children are at particular risk but this risk decreases with increasing age.
Severe seizures, neurological impairment or anticonvulsant therapy may be risk factors. Hepatic failure with pancreatitis increases the risk of fatal outcome.
Patients and guardians should be warned that acute abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical attention. If pancreatitis is diagnosed, sodium valproate should be discontinued and alternative treatment for the underlying medical condition initiated as clinically indicated.
Use in renal impairment.
Lower doses may be required since free drug levels may be high owing to lowered serum albumin and poor urinary excretion of free drug metabolites. As monitoring of plasma concentrations may be misleading, dosage should be adjusted according to clinical monitoring.
Lupus erythematosus.
Although immune disorders have been noted only exceptionally during the use of sodium valproate, the potential benefit of sodium valproate should be weighed against its potential risk in patients with systemic lupus erythematosus.
Hyperammonaemia.
Hyperammonaemia, which may be present in the absence of abnormal liver function tests, can occur in patients during treatment with sodium valproate. This may occasionally present clinically, with or without lethargy or coma, as vomiting, ataxia and increasing clouding of consciousness. Should these symptoms occur, hyperammonaemic encephalopathy should be considered (see Section 4.4 Special Warnings and Precautions for Use, Urea cycle disorders (UCD)) and sodium valproate should be discontinued.
Urea cycle disorders (UCD).
Hyperammonaemic encephalopathy, sometimes fatal, has been reported following initiation of valproate therapy in patients with urea cycle disorders, a group of uncommon genetic abnormalities, particularly ornithine transcarbamylase deficiency.
Prior to the initiation of valproate therapy, evaluation for UCD should be considered in the following patients:
1) those with a history of unexplained encephalopathy or coma, encephalopathy associated with a protein load, pregnancy-related or postpartum encephalopathy, unexplained mental retardation, or history of elevated plasma ammonia or glutamine;
2) those with cyclical vomiting and lethargy, episodic extreme irritability, ataxia, low BUN, or protein avoidance;
3) those with a family history of UCD or a family history of unexplained infant deaths (particularly males);
4) those with other signs or symptoms of UCD.
Patients who develop symptoms of unexplained hyperammonaemic encephalopathy while receiving valproate therapy should receive prompt treatment (including discontinuation of valproate therapy) and be evaluated for underlying urea cycle disorders.
Ornithine transcarbamylase (OTC) deficiency.
The females who are heterozygous for OTC deficiency have a spectrum of clinical and biochemical findings, depending on the extent of inactivation of the X-chromosome. Females may show a range of symptoms due to hyperammonaemia which, may be episodic, and therefore difficult to diagnose. The acute symptoms include headaches, vomiting, irritability, bizarre behaviour, lethargy, ataxia, tremors, seizures (generalised tonic-clonic or focal) and coma.
Valproate may precipitate hyperammonaemia symptoms in those who have pre-existing OTC deficiency. As the symptoms may include seizures, any female with valproate-associated symptomatic hyperammonaemia should be evaluated for OTC deficiency. Investigations should include measurement of plasma amino acids and the immediate cessation of valproate should result in clinical improvement.
Surgery.
Prolongation of bleeding time, sometimes with thrombocytopenia, has occurred with sodium valproate therapy. Platelet function should be monitored before surgery is undertaken in patients receiving sodium valproate.
Other.
Blood tests (blood cell count, including platelet count, bleeding time and coagulation tests) are recommended prior to initiation of therapy or before surgery, and in case of spontaneous bruising or bleeding.
Suicidal behaviour and ideation.
Antiepileptic drugs, including sodium valproate increase the risk of suicidal thoughts or behaviour in patients taking these drugs for any indication. Patients treated with any antiepileptic drug (AED) for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behaviour, and/or any unusual changes in mood or behaviour, and appropriate treatment should be considered.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomised to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behaviour compared to patients randomised to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behaviour or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behaviour for every 530 patients treated.
There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide. The increased risk of suicidal thoughts or behaviour with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behaviour beyond 24 weeks could not be assessed. The risk of suicidal thoughts or behaviour was generally consistent among drugs in the data analysed.
The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analysed. Table 1 shows absolute and relative risk by indication for all evaluated AEDs.
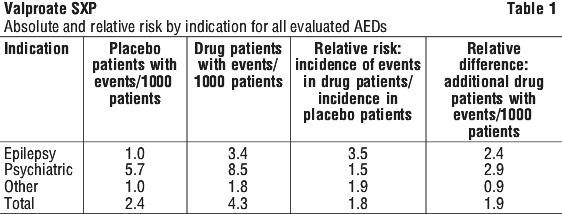 The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications. Anyone considering prescribing sodium valproate or any other AED must balance this risk with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behaviour.
The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications. Anyone considering prescribing sodium valproate or any other AED must balance this risk with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behaviour.
Should suicidal thoughts and behaviour emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behaviour and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behaviour, or the emergence of suicidal thoughts, behaviour, or thoughts about self-harm. Behaviours of concern should be reported immediately to the treating doctor.
Abrupt withdrawal.
The possible risk of fits after sudden cessation of sodium valproate should be borne in mind. If it is the only anticonvulsant used and has to be withdrawn for more than 12 hours because of surgery, control of epilepsy may be lost.
Severe cutaneous adverse reactions and angioedema.
Severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS), erythema multiforme and angioedema, have been reported in association with valproate treatment. Patients should be informed about the signs and symptoms of serious skin manifestations and monitored closely. In case signs of SCARs or angioedema are observed, prompt assessment is needed and treatment must be discontinued if diagnosis of SCARs or angioedema is confirmed.
Carbapenem antibiotics.
The concomitant use of sodium valproate and carbapenem antibiotics is not recommended (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Aggravated convulsion.
As with other antiepileptic drugs, some patients may experience, instead of an improvement, a reversible worsening of convulsion frequency and severity (including status epilepticus), or the onset of new types of convulsions with valproate. In case of aggravated convulsions, the patients should be advised to consult their physician immediately (see Section 4.8 Adverse Effects (Undesirable Effects)).
Thrombocytopenia.
Because of reports of thrombocytopenia, inhibition of the secondary phase of platelet aggregation, and abnormal coagulation parameters, platelet counts and coagulation tests are recommended before initiating therapy and at periodic intervals. Evidence of haemorrhage, bruising or a disorder of haemostasis/coagulation is an indication for reduction of sodium valproate dosage or withdrawal of therapy.
Ornithine transcarbamylase (OTC) deficiency.
A familial history of infant mortality or patient history of OTC deficiency, or of seizures or coma in the presence of mental retardation suggests the need to exclude OTC deficiency.
Weight gain.
Patients should be warned of the risk of weight gain at the initiation of therapy, and appropriate strategies should be adopted to minimise the risk.
Estrogen containing products.
The enzyme inducing effect of valproate is appreciably less than that of certain other anticonvulsants and loss of efficacy of oral contraceptive agents does not appear to be a problem.
However, estrogen containing products, including estrogen containing hormonal contraceptives, may increase the clearance of valproate, which may result in decreased serum concentration of valproate and potentially decreased valproate efficacy. Prescribers should monitor clinical response (seizure control or mood control) when initiating or discontinuing estrogen-containing products. Consider monitoring of valproate serum levels. (See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.)
Use in the elderly.
Although the pharmacokinetics of sodium valproate are modified in the elderly, they have limited clinical significance and dosage should be determined by seizure control. The volume of distribution is increased in the elderly and because of decreased binding to serum albumin, the proportion of free drug is increased. This will affect the clinical interpretation of plasma valproic acid levels.
Paediatric use.
The potential benefit of sodium valproate should be weighed against the risk of pancreatitis or liver damage in such patients prior to initiation of therapy (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). The concomitant use of salicylates should be avoided in children under 3 years of age due to the risk of liver toxicity and the concomitant use of barbiturates may require dosage adjustment (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). Monotherapy is recommended in children under 3 years of age, when prescribing sodium valproate. Young children are at particular risk for pancreatitis, however this risk decreases with increasing age.
The efficacy of sodium valproate for the treatment of manic episodes in bipolar disorder has not been established in patients aged less than 18 years.
Effects on laboratory tests.
Sodium valproate is eliminated mainly through the kidneys, partly in the form of ketone bodies. This may give false positives in the urine testing of possible diabetics.
There have been reports of altered thyroid function test results associated with sodium valproate. The clinical significance of this is unknown.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effects of valproate on other medicines.
Sodium valproate is an inhibitor of a variety of hepatic enzymes, including cytochrome P450, glucuronyl transferase and epoxide hydrolase, and may displace various drugs from plasma protein binding sites. The following list provides information about potential effects of valproate co-administration on a range of commonly prescribed medications. The list is not exhaustive, as new interactions may be reported.
Alcohol.
Valproic acid may potentiate the CNS depressant activity of alcohol. Alcohol intake is not recommended during treatment with valproate.
Antiepileptic drugs.
Several antiepileptic drugs often used in conjunction with valproate (e.g. phenytoin, carbamazepine, phenobarbital (phenobarbitone)) have the ability to increase the intrinsic clearance of valproate, presumably by enzymatic induction of metabolism.
Carbamazepine.
Valproate may displace carbamazepine from protein binding sites and may inhibit the metabolism of both carbamazepine and its metabolite carbamazepine 10, 11 epoxide and consequently potentiate toxic effects of carbamazepine. Clinical monitoring is recommended especially at the beginning of combined therapy, with dosage adjustment when appropriate.
Lamotrigine.
Sodium valproate reduces lamotrigine metabolism and increases its mean half life. This interaction may lead to increased lamotrigine toxicity, in particular serious skin rashes. Clinical monitoring is recommended and lamotrigine dosage should be decreased as appropriate.
Phenobarbital (phenobarbitone).
Sodium valproate blocks the metabolism of barbiturates causing an increase in phenobarbital (phenobarbitone) plasma levels, which, particularly in children, may be associated with sedation. Combination of sodium valproate and phenobarbital (phenobarbitone) can cause CNS depression without significant elevation of serum level of either drug. Therefore, clinical monitoring is recommended throughout the first 15 days of combined treatment. A reduction in the dose of phenobarbital (phenobarbitone) and/or valproate may be necessary and this should also be borne in mind if medicines which are metabolised to phenobarbital (phenobarbitone) (e.g. primidone, methylphenobarbitone) are given with sodium valproate.
Phenytoin.
There have been reports of breakthrough seizures occurring with the combination of sodium valproate and phenytoin. Sodium valproate decreases total plasma phenytoin concentration, however increases in total phenytoin levels have been reported. An initial fall in total phenytoin levels with subsequent increase in phenytoin levels has also been reported. In addition, a decrease in total serum phenytoin with an increase in the free versus protein bound phenytoin levels has been reported with possible overdose symptoms (valproic acid displaces phenytoin from its plasma protein binding sites and reduces its hepatic catabolism). Therefore, clinical monitoring is recommended. When phenytoin plasma levels are determined, the free form should be evaluated. The dosage of phenytoin may require adjustment when given in conjunction with valproate as required by the clinical situation.
Medicines with extensive protein binding.
The concomitant administration of sodium valproate with medicines that exhibit extensive protein binding (e.g. aspirin, carbamazepine, phenytoin, warfarin) may result in alteration of serum drug levels.
Anticoagulants.
The effect of sodium valproate on anticoagulants which modify platelet function is unknown (see Section 4.8 Adverse Effects (Undesirable Effects)). Caution is recommended when administering anticoagulants and other products which have anticoagulant properties (e.g. warfarin and aspirin).
Ethosuximide.
The interaction between ethosuximide and valproate is not normally of clinical significance. There is evidence that sodium valproate may inhibit ethosuximide metabolism, especially in the presence of other anticonvulsants. Patients receiving this combination should be monitored clinically.
Oral contraceptives.
The enzyme inducing effect of valproate is appreciably less than that of certain other anticonvulsants and loss of efficacy of oral contraceptive agents does not appear to be a problem.
Psychotropic agents.
Sodium valproate may potentiate the effects of other psychotropics such as MAOIs, neuroleptics, benzodiazepines and other antidepressants, therefore clinical monitoring is advised and the dose of these medicines should be reduced accordingly.
Clonazepam.
The concomitant use of sodium valproate and clonazepam may produce absence status.
Clozapine.
Caution is advised during concomitant administration as competitive protein binding may potentiate an increase in clozapine or valproate levels.
Diazepam.
Sodium valproate displaces diazepam from its plasma binding sites and inhibits its metabolism. Monitoring of free diazepam levels may be necessary if the patient becomes sedated.
Lorazepam.
A decrease in lorazepam plasma clearance may occur with concomitant administration of sodium valproate.
Midazolam.
Free plasma midazolam may increase in patients receiving valproate. It appears likely that sodium valproate displaces midazolam from its plasma binding sites, potentially leading to an increase of the midazolam response.
Primidone.
Valproate increases primidone plasma levels with exacerbation of its adverse effects (such as sedation); these signs cease with long-term treatment. Clinical monitoring is recommended especially at the beginning of combined therapy with dosage adjustment when appropriate.
Zidovudine.
Valproate may raise zidovudine plasma concentrations leading to increased zidovudine toxicity.
Tricyclic antidepressants.
Sodium valproate may inhibit the metabolism of tricyclic antidepressants. Clinical monitoring of free antidepressant levels may be necessary.
Olanzapine.
Valproic acid may decrease the olanzapine plasma concentration.
Felbamate.
Valproic acid may decrease the felbamate mean clearance.
Rufinamide.
Valproic acid may lead to an increase ion plasma level of rufinamide. This increase is dependent on concentration of valproic acid. Caution should be exercised, in particular in children as this effect is larger in this population.
Propofol.
Valproic acid may lead to an increased blood level of propofol. When co-administered with valproate, a reduction of the dose of propofol should be considered.
Nimodipine.
Concomitant treatment of nimodipine with valproic acid may increase nimodipine plasma concentration.
Other medicines.
There was no notable interaction between valproate and lithium.
Effects of other medicines on valproate.
The dosage of sodium valproate may need to be increased by 5 to 10 mg/kg/day when used in combination with medicines which induce hepatic enzymes and thereby increase the clearance of valproate. In contrast, medicines that are inhibitors of cytochrome P450, may be expected to have only a minor effect on valproate clearance as cytochrome P450 mediated microsomal oxidation is a relatively minor secondary metabolic pathway to glucuronidation and β-oxidation. The list is not exhaustive, as new interactions may be reported.
Aspirin.
Concomitant administration of sodium valproate and aspirin may result in displacement of valproate from protein binding sites, resulting in a rise in free levels. In addition, aspirin appears to inhibit the metabolism of valproate. Thus caution is advisable when patients on sodium valproate are prescribed aspirin. Furthermore, patients requiring long-term aspirin therapy may require a reduction in dosage of sodium valproate.
Felbamate.
Felbamate may decrease valproic acid clearance and consequently increase valproate serum concentrations. Valproate dosage should be monitored when given in combination with felbamate.
Phenobarbital (phenobarbitone), phenytoin and carbamazepine.
These medicines can decrease steady-state valproate levels in patients by increasing the intrinsic clearance of valproate, presumably through enzymic induction of metabolism. The half-life is significantly reduced in patients on polytherapy with these medicines. Dosages should be adjusted according to clinical response and blood levels in case of combined therapy.
Valproic acid metabolite levels may be increased in case of concomitant use with phenytoin or phenobarbital (phenobarbitone). Therefore patients treated with either of these two drugs should be carefully monitored for signs and symptoms of hyperammonemia.
Antidepressants.
Antidepressants (including MAOIs, tricyclic antidepressants and SSRIs) may have the potential to inhibit the metabolism of valproate via the cytochrome P450 system. However, there is not expected to be any significant interaction within normal therapeutic doses. Antidepressants can lower the seizure threshold of non-stabilised epileptic patients, and so careful and regular monitoring of their condition is indicated.
Clozapine.
Caution is advised during concomitant administration as competitive protein binding may potentiate an increase in clozapine or valproate levels.
Chlorpromazine.
Chlorpromazine may inhibit the metabolism of valproate.
Fluoxetine.
Fluoxetine may inhibit the metabolism of valproate as it does with tricyclic antidepressants, carbamazepine and diazepam.
Mefloquine.
Mefloquine increases valproic acid metabolism and has a convulsing effect; therefore epileptic seizures may occur in cases of combined therapy.
Cimetidine or erythromycin.
Valproate serum levels may be increased (as a result of reduced hepatic metabolism) in case of concomitant use with cimetidine or erythromycin.
Carbapenem antibiotics.
Decrease in valproate blood level sometimes associated with convulsions has been observed when valproate and carbapenem antibiotics (panipenem, meropenem, imipenem, ertapenem, biapenem) were combined. Due to the rapid onset and the extent of the decrease, coadministration of carbapenem antibiotics in patients stabilised on valproic acid should be avoided (see Section 4.4 Special Warnings and Precautions for Use).
If treatment with these antibiotics cannot be avoided, close monitoring of valproate blood level should be performed.
Vitamin K dependent factor anticoagulant.
Close monitoring of prothrombin rate should be performed in case of concomitant use of vitamin K dependent factor anticoagulant.
Rifampicin.
Rifampicin may decrease the valproate blood levels resulting in a lack of therapeutic effect. Therefore, valproate dosage adjustment may be necessary when it is co-administered with rifampicin.
Protease inhibitors.
Protease inhibitors such as lopinavir, ritonavir decrease valproate plasma levels when co-administered.
Colestyramine.
Colestyramine may lead to a decrease in plasma levels of valproate when co-administered.
Medicines with extensive protein binding.
The concomitant administration of sodium valproate with medicines that exhibit extensive protein binding (e.g. aspirin, carbamazepine, phenytoin, warfarin) may result in alteration of valproic acid free serum levels.
Estrogen containing products.
Estrogen containing products, including estrogen containing hormonal contraceptives, may increase the clearance of valproate, which may result in decreased serum concentration of valproate and potentially decreased valproate efficacy. Prescribers should monitor clinical response (seizure control or mood control) when initiating or discontinuing estrogen-containing products. Consider monitoring of valproate serum levels.
The enzyme inducing effect of valproate is appreciably less than that of certain other anticonvulsants and loss of efficacy of oral contraceptive agents does not appear to be a problem.
Metamizole.
Metamizole may decrease valproate serum levels when co-administered, which may result in potentially decreased valproate clinical efficacy. Prescribers should monitor clinical response (seizure control or mood control) and consider monitoring valproate serum levels as appropriate.
Methotrexate.
Some case reports describe a significant decrease in valproate serum levels after methotrexate administration, with occurrence of seizures. Prescribers should monitor clinical response (seizure control or mood control) and consider monitoring valproate serum levels as appropriate.
Other interactions.
Risk of liver damage.
The concomitant use of salicylates should be avoided in children under 3 years of age due to the risk of liver toxicity (see Section 4.4 Special Warnings and Precautions for Use, Hepatic dysfunction, Paediatric use).
Concomitant use of valproate and multiple anticonvulsant therapy increases the risk of liver damage, especially in young children (see Section 4.4 Special Warnings and Precautions for Use, Hepatic dysfunction, Paediatric use).
Cannabidiol.
Concomitant use of sodium valproate and cannabidiol at doses 10 to 25 mg/kg may increase the incidence of transaminase enzyme elevations higher than 3 times the ULN (see Section 4.4 Special Warnings and Precautions for Use; Section 4.8 Adverse Effects (Undesirable Effects)). The mechanism of this interaction remains unknown.
Appropriate liver monitoring should be exercised when valproate is used concomitantly with other anticonvulsants with potential hepatotoxicity, including cannabidiol, and dose reductions or discontinuation should be considered in case of significant anomalies of liver parameters (see Section 4.4 Special Warnings and Precautions for Use).
Topiramate and acetazolamide.
Concomitant administration of valproate and topiramate or acetazolamide has been associated with encephalopathy and/or hyperammonemia. Patients treated with those two drugs should be carefully monitored for signs and symptoms of hyperammonemic encephalopathy.
Pivalate-conjugated medicines.
Concomitant administration of valproate and pivalate-conjugated medicines that decrease carnitine levels (such as cefditoren pivoxil, adefovir dipivoxil, pivmecillinam and pivampicillin) may trigger occurrence of hypocarnitinemia (see Section 4.4 Special Warnings and Precautions for Use). Concomitant administration of these medicines with valproate is not recommended. Patients in whom coadministration cannot be avoided should be carefully monitored for signs and symptoms of hypocarnitinemia.
Quetiapine.
Co-administration of valproate and quetiapine may increase the risk of neutropenia/leucopenia.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Amenorrhoea, polycystic ovaries and increased testosterone levels have been reported in women using valproate (see Section 4.8 Adverse Effects (Undesirable Effects)). Valproate administration may also impair fertility in men (see Section 4.8 Adverse Effects (Undesirable Effects)).
In the few cases in which the outcome of switching/discontinuing valproate was reported, the decrease in male fertility potential was reversible in most but not all cases, and successful conceptions have also been observed. There are insufficient data to comment on the effects of reducing the daily dose of valproate in the setting of male infertility.
(Category D)
Treatment of epilepsy. Valproate is contraindicated during pregnancy unless there is no suitable alternative.
Valproate is contraindicated for use in women of childbearing potential, unless the physician has provided education on the potential effects of valproate during pregnancy (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
Treatment of mania. Valproate is contraindicated during pregnancy.
Valproate is contraindicated for use in women of childbearing potential, unless the physician has provided education on the potential effects of valproate during pregnancy (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
Teratogenicity and developmental effects. Valproate was shown to cross the placental barrier both in animal species and in humans (see Section 5.2 Pharmacokinetic Properties).
Animal data.
Teratogenic effects (malformation of multiple organ systems) have been demonstrated in mice, rats, and rabbits.
In published literature, behavioral abnormalities have been reported in first generation offspring of mice and rats after in utero exposure to clinically relevant doses/exposure of valproate. In mice, behavioral changes have also been observed in the 2nd and 3rd generations, albeit less pronounced in the 3rd generation, following an acute in utero exposure of the first generation. The relevance of these findings for humans is unknown.
Pregnancy exposure risk related to valproate. In females, both valproate monotherapy and valproate polytherapy including other antiepileptics, are frequently associated with abnormal pregnancy outcomes. Available data show an increased risk of major congenital malformations and neurodevelopmental disorders in both valproate monotherapy and polytherapy compared to the population not exposed to valproate.
In animals teratogenic effects have been demonstrated in mice, rats and rabbits.
Risk to children of fathers treated with valproate. A retrospective observational study on electronic medical records in 3 European Nordic countries indicates an increased risk of neuro-developmental disorders (NDDs) in children (from 0 to 11 years old) born to men treated with valproate in the 3 months prior to conception compared to those treated with lamotrigine or levetiracetam.
The adjusted cumulative risk of NDDs ranged between 4.0% to 5.6% in the valproate group versus between 2.3% to 3.2% in the composite lamotrigine/levetiracetam monotherapy group. The pooled adjusted hazard ratio (HR) for NDDs overall obtained from the meta-analysis of the datasets was 1.50 (95% CI: 1.09-2.07).
Due to study limitations, it is not possible to determine which of the studied NDD subtypes (autism spectrum disorder, intellectual disability, communication disorder, attention deficit/hyperactivity disorder, movement disorders) contributes to the overall increased risk of NDDs. Alternative therapeutic options and the need for effective contraception while using valproate and for 3 months after stopping the treatment should be discussed with male patients of reproductive potential, at least annually (see Section 4.4 Special Warnings and Precautions for Use).
Congenital malformations from in utero exposure. A meta-analysis (including registries and cohort studies) showed that about 11% of children of epileptic women exposed to valproate monotherapy during pregnancy had major congenital malformations. This is greater than the risk of major malformations in the general population (about 2-3%). The risk of major congenital malformations in children after in utero exposure to anti-epileptic polytherapy including valproate is higher than that of anti-epileptic drugs polytherapy not including valproate. This risk is dose-dependent in valproate monotherapy, and available data suggest it is dose-dependent in valproate polytherapy. However, a threshold dose below which no risk exists cannot be established.
Available data show an increased incidence of minor or major malformations. The most common include: neural tube defects, facial dysmorphism, cleft lip palate, craniostenosis, cardiac, renal and urogenital defects, limb defects (including bilateral aplasia of the radius), and multiple anomalies involving various body systems.
In utero exposure to valproate may also result in hearing impairment/loss due to ear and/or nose malformations (secondary effect) and/or to direct toxicity on the hearing function. Cases describe both unilateral and bilateral deafness or hearing impairment. Outcomes were not reported for all cases. When outcomes were reported, the majority of the cases had not resolved. Monitoring of signs and symptoms of ototoxicity is recommended.
In utero exposure to valproate may result in eye malformations (including colobomas, microphthalmos). These have been reported in conjunction with other congenital malformations. These eye malformations may affect vision.
Data has shown an incidence of congenital malformations in children born to epileptic women exposed to valproate monotherapy during pregnancy. This is a greater risk of major malformations than for the general population. Women treated with sodium valproate have a potentially increased risk of giving birth to a baby with an abnormality due to the higher Cmax of the intravenous formulation compared with the oral formulation.
Mothers taking more than one anticonvulsant medicine might have a higher risk of having a baby with a malformation than mothers taking one medicine.
Sodium valproate (valproic acid), if taken in the first trimester of pregnancy, is suspected of causing an increased risk of neural tube defects (especially spina bifida) in the exposed fetus. This has been estimated to be in the region of 1-2%.
This risk is dose dependent but a threshold dose below which no risk exists cannot be established.
Neurodevelopment disorders from in utero exposure. Data has shown that exposure to valproate in utero can have adverse effects on mental and physical development of the exposed children. The risk of neurodevelopmental disorders (including that of autism) seems to be dose-dependent when valproate is used in monotherapy but a threshold dose below which no risk exists, cannot be established based on data. When valproate is administered in polytherapy with other anti-epileptic drugs during pregnancy, the risks of neurodevelopment disorders in the offspring were also significantly increased as compared with those in children from general population or born to untreated epileptic mothers. The exact gestational period of risk for these effects is uncertain and the possibility of a risk throughout the entire pregnancy cannot be excluded.
When valproate is administered in monotherapy, studies in preschool children exposed in utero to valproate show that some children may experience delays in their early development such as talking and walking later, lower intellectual abilities, poor language skills (speaking and understanding) and memory problems.
Some data have suggested an association between in utero valproate exposure and the risk of developmental delay (frequently associated with craniofacial abnormalities), particularly of verbal IQ. IQ measured in school aged children with a history of valproate exposure in utero, was lower than those children exposed to other antiepileptics. Although the role of confounding factors cannot be excluded, there is evidence in children exposed to valproate that the risk of intellectual impairment may be independent from maternal IQ. There is limited data on the long term outcomes.
Developmental delay has been very rarely reported in children born to mothers with epilepsy. It is not possible to differentiate what may be due to genetic, social, environmental factors, maternal epilepsy or antiepileptic treatment.
Available data from a study conducted using registries in Denmark show that children exposed to valproate in utero are at increased risk of autistic spectrum disorder (approximately 3-fold) and childhood autism (approximately 5-fold) compared to the unexposed population in the study.
Available data from a study conducted using registries in Denmark show that children exposed to valproate in utero are at increased risk of developing attention deficit/hyperactivity disorder (ADHD) (approximately 1.5-fold) compared to the unexposed population in the study.
Lower weight at birth for the gestational age from in utero exposure. In utero exposure to valproate can lead to a lower weight at birth for the gestational age. Available data in humans do not allow to conclude on a potential dose-related effect.
Valproate therapy in pregnancy. The enzyme inducing effect of valproate is appreciably less than that of certain other anticonvulsants and loss of efficacy of oral contraceptive agents does not appear to be a problem.
However, estrogen containing products, including estrogen containing hormonal contraceptives, may increase the clearance of valproate, which may result in decreased serum concentration of valproate and potentially decreased valproate efficacy. Prescribers should monitor clinical response (seizure control or mood control) when initiating, or discontinuing estrogen-containing products. Consider monitoring of valproate serum levels.
Women taking sodium valproate (valproic acid) who become or wish to become pregnant should be encouraged to consider routine ultrasound and amniocenteses for prenatal diagnosis of such abnormalities. As folic acid may have a role in the prevention of neural tube defects in infants of women taking antiepileptic therapy, such women are recommended to take folic acid supplementation (5 mg daily) four weeks prior to and 12 weeks after conception.
Notwithstanding the potential risks, no sudden discontinuation of antiepileptic therapy should be undertaken, without reassessment of the risks and benefits, as this may lead to breakthrough seizures which could have serious consequences for both the mother and the fetus. If after careful evaluation of the risks and benefits, sodium valproate treatment is to be continued during pregnancy, it is recommended to use sodium valproate in divided doses over the day at the lowest effective dose. The use of a prolonged release formulation may be preferable to any other treatment form.
It is recommended that:
in bipolar disorders indication, cessation of valproate therapy should be considered;
women on antiepileptic drugs (AEDs) receive pre-pregnancy counselling with regard to the risk of fetal abnormalities;
AEDs should be continued during pregnancy and monotherapy should be used if possible at the lowest effective dose as risk of abnormality is greater in women taking combined medication;
If appropriate, folic acid supplementation (5 mg) should be commenced four weeks prior to and continue for twelve weeks after conception as it may minimize the risk of neural tube defects;
specialist prenatal diagnosis, including detailed mid-trimester ultrasound, should be offered in order to detect the possible occurrence of neural tube defects or other malformations.
Before sodium valproate is prescribed for use in women with epilepsy of any form, who could become pregnant, they should receive specialist advice. Due to the potential risks to the fetus, the benefits of its use should be weighed against the risks. When treatment with sodium valproate is deemed necessary, precautions to minimise the potential teratogenic risk should be followed (see above recommendations).
If a woman plans a pregnancy.
For the epilepsy indication, if a woman is planning to become pregnant, a specialist experienced in the management of epilepsy must reassess the valproate therapy and consider alternative treatment options. Every effort should be made to switch to appropriate alternative treatment prior to conception, and before contraception is discontinued (see Section 4.4 Special Warnings and Precautions for Use). If switching is not possible, the woman should receive further counselling regarding the valproate risks for the unborn child to support her informed decision making regarding family planning.
For the bipolar disorder indication, if a woman is planning to become pregnant, a specialist experienced in the management of bipolar disorder must be consulted. Treatment with valproate should be discontinued prior to conception, and before contraception is discontinued. If needed, alternative treatment options should be considered.
Folate supplementation (5 mg daily) before the pregnancy may decrease the risk of neural tube defects which may occur in all pregnancies. However the available evidence does not suggest it prevents the birth defects or malformations due to valproate exposure.
Pregnant women.
Valproate as treatment for epilepsy is contraindicated in pregnancy unless there is no suitable alternative treatment (see Section 4.4 Special Warnings and Precautions for Use).
If a woman using valproate becomes pregnant, she must be immediately referred to a specialist to consider alternative treatment options. During pregnancy, maternal tonic clonic seizures and status epilepticus with hypoxia may carry a particular risk of death for mother and the unborn child.
If, despite the known risks of valproate in pregnancy and after careful consideration of alternative treatment, in exceptional circumstances a pregnant woman must receive valproate for epilepsy, it is recommended to:
Use the lowest effective dose and divide the daily dose of valproate into several small doses to be taken throughout the day. The use of a prolonged release formulation may be preferable to other treatment formulations in order to avoid high peak plasma concentrations.
Valproate as treatment for bipolar disorder is contraindicated for use during pregnancy.
All patients with a valproate exposed pregnancy and their partners should be referred to a specialist experienced in pre-natal medicine for evaluation and counselling regarding the exposed pregnancy.
Specialised prenatal monitoring should take place to detect the possible occurrence of neural tube defects or other malformations.
Folate supplementation (5 mg daily) before the pregnancy may decrease the risk of neural tube defects which may occur in all pregnancies. However the available evidence does not suggest it prevents the birth defects or malformations due to valproate exposure.
Risk in the neonate. There have been rare reports of haemorrhagic syndrome in neonates whose mothers have taken sodium valproate during pregnancy. This syndrome is related to thrombocytopenia, hypofibrinaemia, and/or to a decrease in other coagulation factors. Afibrinaemia has also been reported and may be fatal. Hypofibrinaemia is possibly associated with a decrease of coagulation factors. Haemorrhagic syndrome must be distinguished from the decrease of the vitamin-K factors induced by phenobarbital (phenobarbitone) and other enzyme inducers.
Platelet count, fibrinogen plasma level and coagulation status should be investigated in neonates.
Cases of hypoglycaemia have been reported in neonates whose mothers have taken valproate during the third trimester of the pregnancy.
Cases of hypothyroidism have been reported in neonates whose mothers have taken valproate during pregnancy.
Withdrawal syndrome (such as, in particular, agitation, irritability, hyperexcitability, jitteriness, hyperkinesia, tonicity disorders, tremor, convulsions and feeding disorders) may occur in neonates whose mothers have taken valproate during the last trimester of pregnancy.
Sodium valproate is excreted in breast milk. Concentrations in breast milk have been reported to be 1 to 10% of serum concentration. It is not known what effect this would have on a breast-fed infant. As a general rule, breastfeeding should not be undertaken whilst a patient is receiving sodium valproate.4.7 Effects on Ability to Drive and Use Machines
Use of sodium valproate may provide seizure control such that the patient may be eligible to hold a driving license. However, patients should be warned of the risk of drowsiness, especially in cases of anticonvulsant polytherapy, too high a starting dose, too rapid a dose escalation or when used in association with benzodiazepines.
4.8 Adverse Effects (Undesirable Effects)
The adverse events expected for the intravenous formulation of sodium valproate are identical to those expected for the oral formulations currently available with the exception of local administration site reactions.
In epilepsy.
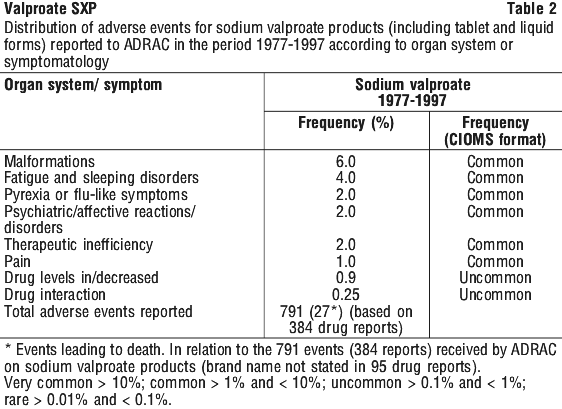
The incidence of adverse reactions to marketed medicines such as sodium valproate is difficult to reliably assess due to the nature of spontaneous reporting systems and the problems associated with estimating the total exposure to the medicine. With these limitations in mind, adverse events received by the Australian Drug Reactions Committee (ADRAC) on sodium valproate products for the twenty-year period 1977-1997 are presented, summarised in Table 2.
The data are presented in accordance with system organ class and include all adverse events reported, independent of drug causality i.e. adverse events classified as certain, probable or possible.
In mania.
No new or unexpected adverse events have been reported in clinical trials of sodium valproate in mania. The frequencies of adverse events (%) reported on valproate (as divalproex) in the largest controlled clinical trial described under Section 5.1 Pharmacodynamic Properties are summarised in Table 3.
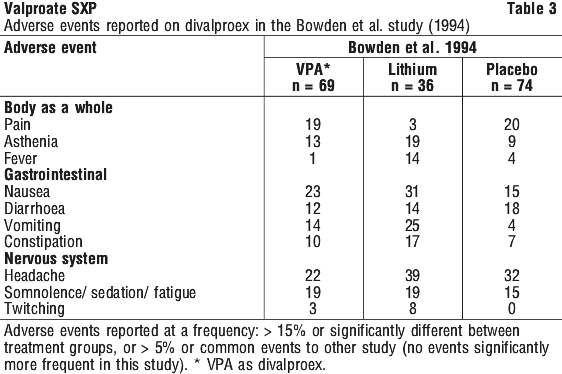 In this study, there were differences with placebo for vomiting only for divalproex (45% vs 14%), fever was more common for lithium (14%) than for divalproex (1%) and placebo (4%), pain was less common with lithium (3%) than with either divalproex (19%) or placebo (20%).
In this study, there were differences with placebo for vomiting only for divalproex (45% vs 14%), fever was more common for lithium (14%) than for divalproex (1%) and placebo (4%), pain was less common with lithium (3%) than with either divalproex (19%) or placebo (20%).
List of adverse effects by system organ class.
Neoplasms benign, malignant and unspecified (including cysts and polyps).
Myelodysplastic syndrome is rare.
Not known: acquired Pelger-Huet anomaly.
Blood and lymphatic system disorders.
Valproic acid inhibits the second stage of platelet aggregation. Reversible prolongation of bleeding time, as well as thrombocytopenia, have been reported but have usually been associated with doses above those recommended (see Section 4.4 Special Warnings and Precautions for Use).
Common cases of thrombocytopaenia and anaemia have been reported.
Uncommon cases of leucopenia and pancytopaenia with or without bone marrow depression have been reported.
Bone marrow failure, including pure red cell aplasia, agranulocytosis, anaemia macrocytic and macrocytosis have rarely been reported.
Isolated cases of decreased blood fibrinogen and prolonged prothrombin time have been reported.
Spontaneous bruising or bleeding is an indication for withdrawal of medication pending investigation (see Section 4.4 Special Warnings and Precautions for Use).
Red cell hypoplasia, neutropenia and leucopenia have also been reported. In most cases the blood picture returned to normal when the medicine was discontinued.
Immune system disorders.
Angioedema, drug rash with eosinophilia and systemic symptoms (DRESS) syndrome and allergic reactions have been observed.
Endocrine disorders.
Hyperandrogenism (hirsutism, virilism, acne, male pattern alopecia, and/or androgen increased) and syndrome of inappropriate secretion of ADH (SIADH) is uncommon.
Hypothyroidism is rare.
Metabolism and nutrition disorders.
Common cases of hyponatremia have been reported. Increased weight is common and since this is a risk factor for polycystic ovary syndrome, it should be carefully monitored.
Obesity has been reported rarely.
Hyperammonaemia is rare. This has been reported in association with valproate therapy and may be present despite normal liver function tests.
Hyperammonaemia associated with neurological symptoms has been reported. In patients who develop unexplained lethargy and vomiting or changes in mental status, further investigations and hyperammonaemic encephalopathy should be considered. In these patients, EEG and ammonia level should be checked and, if ammonia is increased, valproate therapy should be discontinued. Appropriate interventions for treatment of hyperammonaemia should be initiated, and such patients should undergo investigation for underlying urea cycle disorders (see Section 4.4 Special Warnings and Precautions for Use).
Not known: hypocarnitinemia (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
Asymptomatic elevations of ammonia are more common and, when present, require close monitoring of plasma ammonia levels. If the elevation is significant (above 3N) and persists, discontinuation of valproate therapy should be considered.
Psychiatric disorders.
Confusional state, hallucinations, aggression, agitation, disturbance in attention, abnormal behaviour, psychomotor hyperactivity and learning disorder have been commonly reported.
Paediatric population.
The safety profile of valproate in the paediatric population is comparable to adults, but some adverse reactions are more severe or principally observed in the paediatric population. There is a particular risk of severe liver damage in infants and young children especially under the age of 3 years. Young children are also at particular risk of pancreatitis. These risks decrease with increasing age (see Section 4.4 Special Warnings and Precautions for Use). Psychiatric disorders such as aggression, agitation, disturbance in attention, abnormal behaviour, psychomotor hyperactivity and learning disorder are principally observed in the paediatric population.
Nervous system disorders.
The true incidence of drowsiness and sedation with sodium valproate is difficult to assess, as mostly it was administered in combination with other medicines. Sodium valproate, however, may have an intrinsic sedative action in addition to potentiating sedative effects of other anticonvulsants (e.g. barbiturates, clonazepam) and CNS depressants, including alcohol.
Very common cases of tremor have been reported.
Common cases of stupor, somnolence, convulsion, memory impairment, headache, nystagmus and dizziness have been reported.
When using sodium valproate intravenously, dizziness may occur a few minutes after injection; it disappears spontaneously within a few minutes.
Common cases of extrapyramidal disorder which may not be reversible, including reversible parkinsonism has been reported.
Uncommon cases of ataxia, coma, encephalopathy, aggravated convulsions (see Section 4.4 Special Warnings and Precautions for Use), lethargy, paresthesia and reversible parkinsonism have been reported.
Diplopia and depression have occurred rarely and usually in association with other anticonvulsants. Excitement, hyperactivity and behavioural disorders have been rarely reported, usually in children at the start of treatment.
Rare cases of reversible dementia associated with reversible cerebral atrophy and cognitive disorder have been reported.
Stupor and lethargy sometimes leading to transient coma/encephalopathy, either isolated or in conjunction with recurrence of seizures, may occur and were most often associated with polytherapy, too high a starting dose or too rapid a dose escalation.
Eye disorders.
Not known: diplopia.
Ear and labyrinth disorders.
Deafness, either reversible or irreversible, has been reported commonly.
Vascular disorders.
Haemorrhage is common and the occurrence of vasculitis is uncommon.
Respiratory, thoracic and mediastinal disorders.
Pleural effusion has uncommonly been reported.
Pneumonia.
Gastrointestinal disorders.
Nausea is very common.
Vomiting is common. Upper abdominal pain, diarrhoea, gingival disorder (mainly gingival hyperplasia) and stomatitis are common and frequently occur at the start of treatment and usually disappear after a few days without discontinuing treatment.
Vomiting, abdominal cramp, upper abdominal pain, anorexia, increased appetite and diarrhoea are usually transient and rarely require discontinuation of therapy or limitation of dose.
The overall incidence of adverse GI effects are reported to be 9 to 16% in adults and over 22% in children when plain tablets are prescribed. GI side effects may be minimised by taking the tablets with or after food or by substituting the enteric-coated tablets. As some of these symptoms may also indicate early stage hepatic dysfunction, patients should be monitored closely for the appearance of these symptoms. Patients should be instructed to report such signs to the clinician for investigation should they occur (see Section 4.4 Special Warnings and Precautions for Use).
There have been uncommon reports of pancreatitis, sometimes lethal, occurring in patients receiving valproic acid or sodium valproate, usually within the first 6 months of therapy.
Patients experiencing acute abdominal pain should have the serum amylase estimated promptly; if these levels are elevated the medicine should be withdrawn (see Section 4.4 Special Warnings and Precautions for Use).
Hepatobiliary disorders.
Liver injury is common.
Hepatic dysfunction, including hepatic failure resulting in fatalities, has occurred in patients whose treatment included valproic acid or sodium valproate (see Section 4.4 Special Warnings and Precautions for Use).
Concomitant valproate and cannabidiol can cause elevations of ALT and AST greater than 3 times the upper limit of normal (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Skin and subcutaneous tissue disorders.
Hypersensitivity and transient and/or dose related alopecia has been commonly observed. This effect does not appear to be dose-related and regrowth may occur, although the hair may become more curly than previously.
Nail and nail bed disorders have been commonly reported.
Hirsutism, acne and male pattern alopecia are uncommon (see Section 4.8 Adverse Effects (Undesirable Effects), List of adverse effects by system organ class, Endocrine disorders). Angioedema, rash and hair disorder (such as hair texture abnormal, hair colour changes, hair growth abnormal) are uncommon.
Toxic epidermal necrolysis, erythema multiforme, drug rash with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome have been rarely reported.
Dermatological reactions consistent with immune adverse reactions such as pruritus, urticaria have been noted. Caution should be observed when using the medicine in patients with systemic lupus erythematosus.
Not known: hyperpigmentation.
Musculoskeletal and connective tissue disorders.
Decreased bone mineral density, osteopenia, osteoporosis and fractures in patients on long-term therapy with valproate have been uncommon. The mechanism by which valproate affects bone metabolism has not been identified.
Systemic lupus erythematosus and rhabdomyolysis are rare.
Renal and urinary disorders.
Urinary incontinence is common.
Renal failure is uncommon.
Rare cases of enuresis and tubulointerstitial nephritis have been reported.
Rare cases of reversible Fanconi's syndrome associated with valproate therapy have been reported but the mode of action is as yet unclear.
Reproductive system and breast disorders.
Dysmenorrhoea is common and amenorrhoea is uncommon.
There have been rare reports of male infertility and polycystic ovaries.
There have been reports of irregular menses and secondary amenorrhoea and rare cases of breast enlargement and galactorrhoea.
General disorders and administration site conditions.
Non-severe peripheral oedema and hypothermia are uncommon.
Oedema has been reported. Increase in appetite may occur.
Investigations.
Coagulation factors decreased, abnormal coagulation tests (such as prolonged prothrombin time, activated partial thromboplastin time, thrombin time and INR) and biotin deficiency/biotinidase deficiency have rarely been reported.
Reporting of suspected adverse reactions.
Reporting of suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems (Australia) or https://nzphvc.otago.ac.nz/reporting/ (New Zealand).4.9 Overdose
Cases of accidental and suicidal overdosage have been reported. Fatalities are rare.
Symptoms.
Symptoms of overdosage may include serious CNS depression and impairment of respiration. In cases of overdose, long half-lives up to 30 hours have been reported. Signs of an acute massive overdose usually include coma, with muscular hypotonia, hyporeflexia and miosis, impaired respiratory functions and metabolic acidosis, hypotension and circulatory collapse/shock. Symptoms may however be variable and seizures have been reported in the presence of very high plasma levels. Cases of intracranial hypertension related to cerebral oedema have been reported. Deaths have occurred following massive overdose; nevertheless, a favourable outcome is usual. The presence of sodium content in the valproate formulations may lead to hypernatraemia when taken in overdose.
Treatment.
Establish airway and breathing and evaluate circulatory status. Assisted mechanical ventilation may be required in cases of respiratory depression. Haemodialysis and haemoperfusion have been used successfully. Intravenous naloxone has also been used sometimes in association with activated charcoal given orally when used in cases of overdosage with oral formulations. In case of valproate overdose resulting in hyperammonemia, carnitine can be given through IV route to attempt to normalize ammonia levels.
Provided that adequate supportive treatment is given, full recovery usually occurs. Particular attention should be given to the maintenance of an adequate urinary output. Hepatic and pancreatic function should be monitored.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia) or the National Poisons Centre on 0800 POISON or 0800 764 766 (New Zealand).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antiepileptics, ATC Code: N03AG01.
Class: Anticonvulsant, antipsychotic.
Mechanism of action.
Site and mode of action.
The mode of action of sodium valproate has not been fully established. Its anticonvulsant effect is attributed to the blockade of voltage dependent Na+ channels and increased brain levels of γ-aminobutyric acid (GABA). The GABA-ergic effect is also believed to possibly contribute towards the antimanic properties of sodium valproate.
In animals, sodium valproate raises cerebral and cerebellar levels of the inhibitory synaptic transmitter, GABA, possibly by inhibiting GABA degradative enzymes, such as GABA transaminase and/or succinic semialdehyde dehydrogenase and/or by inhibiting the reuptake of GABA by neuronal cells.
Sodium valproate exhibits marked anticonvulsant activity in animals, demonstrated by the various tests used to detect antiepileptic activity.
Sodium valproate appears to have no significant hypnotic effect (an incidence of about 0.2% was noted for drowsiness in a survey of unwanted effects), nor does it have any significant action on the autonomic nervous system, respiration, blood pressure, renal function or body temperature. It does have a spasmolytic action on the isolated ileum preparation but no effect on the nictitating membrane.
Pharmacodynamics.
In epilepsy.
Sodium valproate has been shown to be effective in the treatment of absence seizures (petit mal), tonic-clonic seizures (grand mal) and myoclonic seizures. It has also been shown to be effective in patients with partial (focal) seizures. Sodium valproate appears to have less sedative effect than conventional antiepileptic drugs and this, together with the reduction in fit frequency in children, has often led to improvements in alertness and performance in school.
In mania.
In one study valproate has been shown to be significantly more effective than placebo in the treatment of acute mania and has been reported to be comparable to lithium. Potential medicine interactions likely to be relevant to valproate in the management of patients with mania are outlined under Section 4.5 Interactions with Other Medicines and Other Forms of Interactions. Although the dosage of sodium valproate varied considerably among the controlled studies, a fixed initial dose was used after which dosage was determined by serum levels.
Clinical trials.
In epilepsy.
Sodium valproate efficacy in this therapeutic indication is widely known and recognised.
In mania.
There have been at least five double-blind trials comparing sodium valproate or the bioequivalent active, divalproex sodium with either placebo and/or lithium in the treatment of mania. Only one of these trials was of adequate size. Bowden et al (1994) demonstrated most convincingly the superior effectiveness of valproate as compared to placebo in the treatment of acute mania. Marked improvement, defined as at least 50% improvement on the Manic Syndrome Subscale of the Mania Rating Scale occurred in 48% of valproate-treated patients and 25% of placebo-treated patients respectively (p=0.0040). Comparable efficacy to lithium in this study was reported. Marked improvement, defined as at least 50% improvement on the Manic Syndrome Subscale of the Mania Rating Scale, occurred in a similar number of patients receiving sodium valproate and lithium, 48% and 49% respectively.
5.2 Pharmacokinetic Properties
Absorption.
The pharmacokinetic profile of sodium valproate injection differs from that of oral sodium valproate preparations. As expected, after intravenous and oral dosage (enteric-coated tablets) of sodium valproate (400 mg), Tmax is reached sooner following intravenous administration (7.3 ± 2.6 min) than after oral administration (227.7 ± 59.2 min) and Cmax is higher after intravenous dosage (55.4 ± 9.38 microgram/mL) than after oral administration (39.1 ± 3.51 microgram/mL). The bioavailability of sodium valproate enteric-coated (EC) tablets is only slightly less than that of intravenous sodium valproate with a mean AUC ratio of 100:87 for intravenous to oral forms respectively. The distribution, metabolism, excretion and elimination of intravenous sodium valproate are not different to orally administered sodium valproate.
In most adult patients, daily doses of 1,200 to 1,500 mg result in therapeutic plasma levels of 50 to 100 microgram/mL (0.35 to 0.69 mmol/L). However, correlation between the daily dose per bodyweight and plasma levels of drug has been poor.
Distribution.
Distribution of sodium valproate is rapid and most likely restricted to the circulation and rapidly exchangeable extracellular water. CSF and breast milk levels were found to be 5 to 15% and about 1 to 10% of plasma levels, respectively.
Valproic acid shows non-linear kinetics, due to concentration-dependent plasma protein binding as well as a relatively short half-life.
Sodium valproate is approximately 90% bound to plasma proteins but only 60% to albumin. However, if the plasma level of valproic acid rises above 120 microgram/mL or if the serum albumin concentration is lowered, the binding sites may become saturated, causing the amount of free drug to rise rapidly, out of proportion to any increase in dosage. Sodium valproate may displace phenobarbital (phenobarbitone) or phenytoin from plasma protein binding sites.
Saliva levels of sodium valproate are poorly correlated with those in plasma in contrast to the good correlation found for other antiepileptics.
Placental transfer (see Section 4.6 Fertility, Pregnancy and Lactation). Valproate crosses the placental barrier in animal species and in humans:
In animal species, valproate crosses the placenta, to a similar extent as in humans.
In humans, several publications assessed the concentration of valproate in the umbilical cord of neonates at delivery. Valproate serum concentration in the umbilical cord, representing that in the fetuses, was similar to or higher than that in the mothers.
Metabolism.
Its metabolism is complex; the major elimination pathway is via glucuronidation (40-60%). The remainder is largely metabolised via oxidation pathways, β-oxidation accounting for 30-40% and w-oxidation (cytochrome P450 dependent), the remaining fraction. Only 1 to 3% of the ingested dose is found to be excreted unchanged in the urine.
Excretion.
Sodium valproate is almost completely metabolised prior to excretion. Plasma half-life is variable but generally appears to be 8 to 12 hours (range 3.84 to 15.77 hours) in adults. It may be shorter in patients receiving other anticonvulsants and patients receiving the medicine for long periods. In cases of overdose, long half-lives up to 30 hours have been reported.
Antipsychotic agents or antidepressants including MAOIs, tricyclics and SSRIs co-administered with sodium valproate may result in competitive metabolism or enzyme inhibition, thereby increasing valproate levels (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Children and adolescents.
In paediatric patients below the age of 10 years, the systemic clearance of valproate varies with age. In neonates and infants up to 2 months of age, valproate clearance is decreased when compared to adults and is lowest directly after birth. In a review of the scientific literature, valproate half-life in infants under two months showed considerable variability ranging from 10 to 67 hours. In children aged 2-10 years, valproate clearance is 50% higher than in adults. Above the age of 10 years, children and adolescents have valproate clearances similar to those reported in adults.
5.3 Preclinical Safety Data
Genotoxicity.
Valproate was not mutagenic in bacteria (Ames test), or mouse lymphoma L5178Y cells at thymidine kinase locus (mouse lymphoma assay), and did not induce DNA repair activity in primary culture of rat hepatocytes. After oral administration, valproate did not induce either chromosome aberrations in rat bone marrow, or dominant lethal effects in mice.
Statistically significant higher incidences of sister-chromatid exchange (SCE) have been observed in patients exposed to valproate as compared to healthy subjects not exposed to valproate. However, these data may have been impacted by confounding factors. Two published studies examining SCE frequency in epileptic patients treated with valproate versus untreated epileptic patients, provided contradictory results. The biological significance of an increase in SCE frequency is not known.
Toxicology.
No significant toxic effects were seen in rats receiving 270 mg/kg/day for 3 months or in dogs receiving 90 mg/kg/day for 12 months. At higher doses sedation, ataxia and various histopathological effects (testicular atrophy and reduction in lymphoid tissue) were observed at levels of 256 to 568 microgram/mL (1.78 to 3.94 mmol/L).
Testicular function.
In sub-chronic/chronic toxicity studies, testicular degeneration/atrophy or spermatogenesis abnormalities and a decrease in testes weight were reported in adult rats and dogs after oral administration of valproate. The dose without an effect on the testes was similar to the maximum recommended human dose of 50 mg/kg/day on a mg/m2 basis.
In a fertility study in rats, valproate at doses up to 350 mg/kg/day did not alter male reproductive performance. This dose was about 1.3 times the maximum recommended human dose of 50 mg/kg/day on a mg/m2 basis.
In juvenile rats, a decrease in testes weight and testicular degeneration were observed at doses that also elicited substantial general systemic toxicity. The relevance of the testicular findings to the paediatric population is unknown because the fertility of animals exposed to valproate as pre-pubertal juveniles has not been investigated.
Carcinogenicity.
Carcinogenesis.
Sodium valproate was administered in the diet to Sprague-Dawley rats and ICR (HA/ICR) mice at approximate dosage levels of 0, 80 and 160 mg/kg/day for up to 2 years. There was equivocal evidence of an increased incidence of subcutaneous fibrosarcomas in male rats and of bronchoalveolar adenomas in male mice. The presence of these tumours was not considered to be biologically significant because of the published variable incidence of spontaneously occurring fibrosarcomas and pulmonary adenomas in rats and mice respectively and the fact that statistical significance of tumour incidence was only attained in males. The significance of these findings for humans is unknown at present.6 Pharmaceutical Particulars
6.1 List of Excipients
Disodium edetate, hydrochloric acid, sodium hydroxide, water for injections.
6.2 Incompatibilities
Incompatibilities were either not accessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store in a dry place below 30°C.
To reduce microbiological hazard, use as soon as practicable after dilution. If storage is necessary, hold at 2 to 8°C for not more than 24 hours.
6.5 Nature and Contents of Container
300 mg/3 mL solution for injection/infusion.
Valproate SXP, solution for injection/infusion is packaged in a 10 mL, Type I glass vial, stoppered with a Teflon coated bromobutyl rubber stopper and an aluminium flip-off cap.
Pack size: 5 x 10 mL vial.
400 mg/4 mL solution for injection/infusion.
Valproate SXP, solution for injection/infusion is packaged in a 10 mL, Type I glass vial, stoppered with a Teflon coated bromobutyl rubber stopper and an aluminium flip-off cap.
Pack size: 5 x 10 mL vial.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Sodium valproate is a white, odourless, crystalline powder with a saline taste. It is highly soluble in water and alcohol. It is quite dissimilar to other established anticonvulsants such as barbiturates, hydantoins, succinamides, oxazolidinediones and acetylureas in that it has no nitrogen or aromatic moiety.
Chemical name: Sodium di-n-propylacetic acid.
Molecular weight: 166.2.
Molecular formula: C8H15NaO2.
Chemical structure.

CAS Number.
1069-66-5.7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine (S4).
Summary Table of Changes
