1. Why am I using VEKLURY?
VEKLURY contains the active ingredient remdesivir.
VEKLURY is an anti-virus medicine.
VEKLURY is used to treat COVID-19.
COVID 19 is caused by a virus called a coronavirus. VEKLURY stops the virus in cells from reproducing, and this stops the virus multiplying in the body. This can help your body to overcome the virus infection and may help you get better faster.
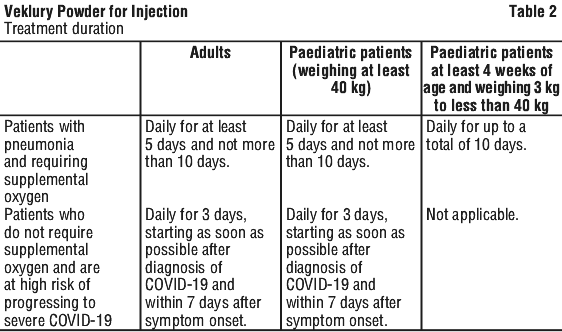
VEKLURY will be given in hospital to some people with COVID 19. It is suitable for adults and adolescents (aged 12 years and over) weighing at least 40 kg. It will only be given to patients who have pneumonia, and need extra oxygen to help them breathe. It will be given to you by a doctor or nurse, as a drip into a vein (an intravenous infusion), lasting 30 to 120 minutes, once a day. You will be given VEKLURY every day for at least 5 days. Your doctor may extend the treatment up to a total of 10 days. You will be closely monitored during your treatment.
2. What should I know before I use VEKLURY?
Warnings
Do not use VEKLURY if:
- You are allergic to remdesivir, or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any other medical conditions
- take any medicines for any other condition
- if you have liver problems. Some people developed increased liver enzymes when given VEKLURY. Your doctor will do blood tests before starting treatment to check whether you can be given it safely.
- have kidney problems. Some people with severe kidney problems may not be given this medicine. Your doctor will do blood tests to check whether you can be given it safely.
- if you are pregnant or breast-feeding. Talk to your doctor or nurse if you are pregnant (or you might be), or if you are breast-feeding.
Reactions following the infusion
VEKLURY can cause allergic reactions or reactions following the infusion. Symptoms can include:
- Changes to blood pressure or heart rate.
- Low oxygen level in blood
- High temperature
- Shortness of breath, wheezing
- Swelling of the face, lips, tongue or throat (angioedema)
- Rash
- Feeling sick (nausea)
- Sweating
- Shivering.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.This is because the effects of VEKLURY in pregnant or breastfeeding women are not known, and it may harm your unborn baby or your breastfeeding child. VEKLURY will only be given if the potential benefits of treatment outweigh the potential risks to the mother and the unborn baby.
It is not yet known whether VEKLURY or the COVID 19 virus pass into human breast milk, or what the effects might be on the baby or milk production. Your doctor will help you decide whether to continue breast-feeding or to start treatment with VEKLURY. You will need to consider the potential benefits of treatment for you, compared with the health benefits and risks of breast-feeding for your baby.
Blood tests before and during treatment
If you are prescribed VEKLURY, you will be given blood tests before treatment starts. Patients being treated with VEKLURY will have blood tests every day during their treatment. These tests are to check for kidney or liver problems. VEKLURY will be stopped if your kidney or liver show signs of damage during treatment.
Children and adolescents
VEKLURY is suitable for adults and adolescents (aged 12 years and over) weighing at least 40 kg.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Do not take chloroquine or hydroxychloroquine at the same time as VEKLURY.
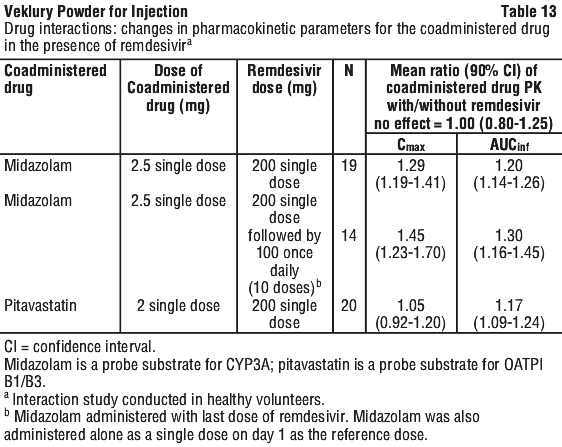
Certain medicines e.g. midazolam or pitavastatin should be taken at least 2 hours after VEKLURY as VEKLURY can affect the way they work.
VEKLURY may affect the way certain medicines (e.g. theophylline or midazolam) work.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect VEKLURY.
VEKLURY can be used with dexamethasone.
It is not yet known if VEKLURY affects other medicines or is affected by them. Your doctor will monitor you for signs of medicines affecting each other.
4. How do I use VEKLURY?
How much to take / use
VEKLURY be given to you in hospital, by a nurse or doctor, as a drip into a vein (an intravenous infusion) lasting 30 to 120 minutes, once a day. You will be closely monitored during your treatment.
The recommended dose is
- a single starting dose of 200 mg on day 1
- then daily doses of 100 mg starting on day 2.
How long a course of treatment lasts depends on how unwell you are:
- You will be given VEKLURY every day for at least 5 days. Your doctor may extend the treatment up to a total of 10 days.
If you forget to use VEKLURY
As VEKLURY is only used in hospital, it is very unlikely that you will miss a dose. If you have missed one tell your doctor immediately.
If you use too much VEKLURY
As VEKLURY is only used in hospital, it is very unlikely that you will receive too much. If you have been given an extra dose tell your doctor immediately.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using VEKLURY?
Blood tests before and during treatment.
If you are prescribed VEKLURY, you will be given blood tests before treatment starts. Patients being treated with VEKLURY will have blood tests every day during their treatment. These tests are to check for kidney or liver problems. VEKLURY will be stopped if your kidney or liver show signs of damage during treatment.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how VEKLURY affects you. VEKLURY is not expected to have any effect on your ability to drive.
Looking after your medicine
This medicine will usually be stored in the hospital pharmacy.
Powder for injection:
- Before use, this medicinal product does not require any special storage conditions.
- Once reconstituted, VEKLURY should be diluted immediately.
- Once diluted, VEKLURY should be used immediately. If necessary, bags of diluted solution can be stored for up to 4 hours at room temperature (20°C to 25°C), or for up to 24 hours in a fridge (2°C to 8°C). Do not allow more than 24 hours between dilution and administration.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.

Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell. Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effect you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only used in hospital.
What VEKLURY contains

Do not take this medicine if you are allergic to any of these ingredients.
What VEKLURY looks like
VEKLURY 100 mg powder for injection is a white, off-white to yellow powder, to be reconstituted and then diluted into 0.9% saline prior to administration by intravenous infusion. It is sterile, preservative-free, and supplied in a single-use clear glass vial.
(AUST R 338419)
VEKLURY is available in cartons containing 1 vial.
Who distributes VEKLURY
Gilead Sciences Pty Ltd
Level 6, 417 St Kilda Road
Melbourne, Victoria 3004
This leaflet was prepared in July 2020.
VEKLURY and GILEAD are registered trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
Published by MIMS September 2020

 Veklury is to be administered via IV infusion over 30 to 120 minutes.
Veklury is to be administered via IV infusion over 30 to 120 minutes.




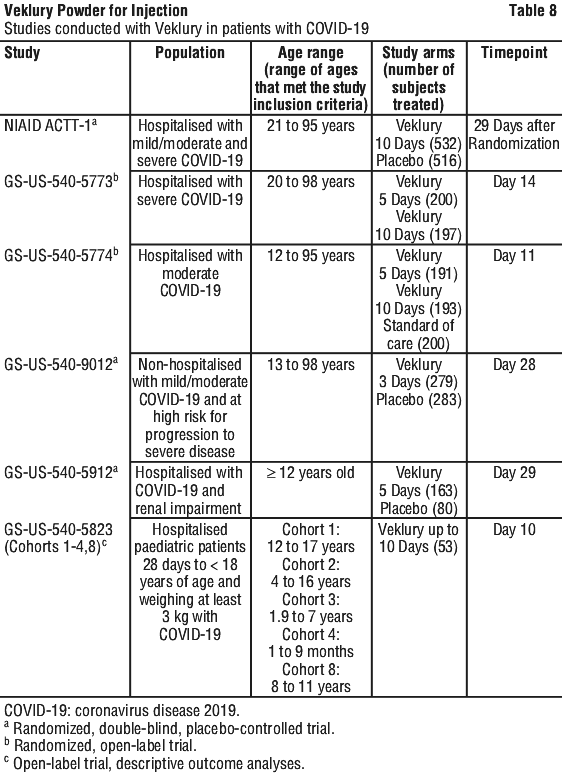 These studies were conducted in a population that had not been vaccinated against COVID-19.
These studies were conducted in a population that had not been vaccinated against COVID-19.