What is in this leaflet
This leaflet answers some common questions about VELETRI. It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What VELETRI is used for
VELETRI is used to treat some types of pulmonary arterial hypertension (PAH). PAH is characterised by high blood pressure in the blood vessel that carries blood from the heart to the lungs, and increased resistance in the blood vessels of the lung. The cause of PAH is not known however there are a number of diseases such as scleroderma that are associated with PAH. VELETRI belongs to a group of medicines called prostaglandins.
VELETRI works by widening the blood vessels in the lungs and so lowering the blood pressure in your lungs (known as a vasodilator action).
Your doctor however, may prescribe VELETRI for another purpose.
Ask your doctor if you have any questions about why it has been prescribed for you.
VELETRI is not addictive.
Before you use VELETRI
When you must not use VELETRI
Do not use VELETRI if you have ever had an allergic reaction to epoprostenol or any of the ingredients listed at the end of this leaflet. Symptoms of an allergic reaction may be mild or severe. They usually include some or all of the following: wheezing, swelling of the lips/mouth, difficulty in breathing, hayfever, lumpy rash ("hives") or fainting.
Do not use VELETRI if you have heart disease with shortness of breath, and swelling of the feet or legs due to fluid build-up.
Do not use VELETRI after the expiry date [EXP.] printed on the pack. If you use it after the expiry date has passed, it may not work as well.
Do not use VELETRI if the packaging is torn or shows signs of tampering.
If you're not sure whether you should be using VELETRI, talk to your doctor.
Before you start to use VELETRI
You must tell your doctor if:
- you are allergic to foods, dyes, preservatives or any other medicines.
- you have heart disease
- you are pregnant, trying to become pregnant, or breastfeeding.
- you are taking any medicine to prevent blood clots, such as heparin, warfarin, aspirin or other anti-inflammatory pain killers (NSAIDs).
- you are taking any medicines that are used to treat high blood pressure, or a group of medicines known as nitrates that are used to treat angina.
- you are taking digoxin, a medicine used to treat heart failure.
- you are taking any other medicines, including medicines you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines may affect the way others work. Your doctor or pharmacist will be able to tell you what to do when using VELETRI with other medicines.
If you develop pulmonary oedema (water on the lungs) during this time, your doctor may choose not to treat you with VELETRI.
How to use VELETRI
VELETRI will be given as an intravenous infusion only, normally through a permanently fitted intravenous catheter (during initial treatment a 'peripheral line' may be used which is a non-permanent catheter). Before VELETRI is used, it must be dissolved only in sterile water for injection or Sodium Chloride 0.9% solution for injection.
How much to use
Initial treatment with VELETRI will be carried out in a hospital. Your doctor will start you on an infusion and slowly increase the dose (every 15 minutes) to find the most effective or highest dose you can tolerate.
During this part of the treatment you will also learn about how your body reacts to VELETRI.
Your doctor will then continue the infusion based on this dose, and may increase or decrease your infusion rate depending on your response to the treatment. All changes should be done gradually and under the direction of a doctor, except in emergency situations.
How to use VELETRI
Your VELETRI infusion will be given to you as continuous intravenous infusion only, normally through a permanently fitted intravenous catheter through a pump. There are only certain pumps which can be used. Your doctor will make sure you are using the right one.
Your doctor or nurse will have shown you how to keep your catheter clean, and the area around it clean and free from infection. They will also show you how to prepare and administer VELETRI and how to stop treatment if necessary. It is very important you follow their instructions carefully.
VELETRI contains no preservative. Use a vial once only and then discard.
How long to use it
Use VELETRI for as long as your doctor advises you to. VELETRI is generally used over a prolonged period of time, possibly years. It should not be stopped suddenly. Symptoms of suddenly stopping VELETRI include dizziness, weakness and difficulty breathing.
If you use too much (overdose)
As VELETRI has vasodilatory action, overdose may lead to low blood pressure, loss of consciousness, nausea, diarrhoea, vomiting, facial flushing, headache and fast heart beat.
In hospital, the effects of VELETRI are monitored carefully by your doctor. In the unlikely event that you receive too much, appropriate action, such as reducing the dose can be taken promptly.
If you are using VELETRI at home and you think you have used too much, immediately telephone your doctor or the nearest hospital casualty department, even if there are no signs of discomfort. You may need urgent medical attention.
Poison Information Centre telephone numbers:
- Australia: 13 11 26
- New Zealand: 0800 POISON or 0800 764 766
Keep telephone numbers for these places handy.
While you are using VELETRI
Things you must do
Tell your doctor or pharmacist that you are using VELETRI if you are about to start on any new medicines.
Tell your doctor if you become pregnant or are trying to become pregnant.
Tell your doctor if you are breastfeeding or plan to breastfeed.
Tell your doctor if you have buildup of fluid in the abdomen causing pain and swelling.
Tell your doctor if, for any reason, you have not used your medicine exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
It is very important to keep the area around the intravenous catheter clean, otherwise infection of the skin at the site of injection may result, which can then spread into your blood (known as septicaemia).
During administration of VELETRI the intravenous catheter may become blocked. Tell your doctor or pharmacist if this happens.
Things you must not do
Do not stop using or change the dose without first checking with your doctor.
Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
Do not use VELETRI to treat any other complaints unless your doctor says to.
Things to be careful of
Be careful driving or operating machinery until you know how VELETRI affects you. If you are affected, do not drive or operate machinery. As with many other medicines, VELETRI may cause dizziness/ drowsiness/tiredness in some people.
Side Effects
Check with your doctor as soon as possible if you have any problems while taking VELETRI, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Like other medicines, VELETRI can cause side effects in some people.
If you think you are having an allergic reaction to VELETRI while you are receiving it, TELL YOUR DOCTOR IMMEDIATELY.
Symptoms usually include some or all of the following:
- wheezing
- swelling of the lips/mouth
- difficulty in breathing
- hay fever
- lumpy rash ("hives")
- fainting
Tell your doctor at once if you experience any of the following while you are receiving VELETRI:
- fever
- fatigue
- chills/flu-like symptoms
- facial flushing or paleness
- fast heart beat
- slow heart rate
- chest pain and tightness
- heart attack
- shortness of breath
- diarrhoea
- feeling sick (nausea) or being sick (vomiting)
- wind or tummy discomfort
- anxiety, nervousness and agitation
- depression/ psychotic depression
- acute confusional state
- dizziness, especially on standing
- headaches
- dry mouth
- itchy skin rash (eczema) or hives
- decreased or increased feeling or sensitivity, especially in the skin
- tingling or numbness of the hands or feet
- reddening and pain at the infusion site
- sweating
- pain
- jaw, muscle and/or back pain
- joint pain
- loss of appetite
- low number of platelets or red blood cells or all types of blood cells
- bleeding: any bleeding can be serious, if this occurs you should contact your doctor
- ulcer (sore) on the skin
- lung infection
- fluid collection in the chest
- feeling very tired
- blood clot that moves to the lungs, causes chest pain and makes you short of breath
- air leaks into the space between lung and chest wall (collapsed lung)
- muscle tremors
- increased body movement
- overactive thyroid
- urinary tract infection
- increase size of the spleen
Do not be alarmed by this list of possible side effects.
You may not experience any of them.
This is not a complete list of all possible side-effects. Others may occur in some people and there may be some side-effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor if you don't understand anything in this list.
VELETRI may affect your blood sugar levels, heart rate or blood pressure during infusion. Your doctor will monitor these.
Storing VELETRI
Storage
Keep this medicine where young children cannot reach it. A locked cupboard at least one-and-a half metres above the ground is a good place to store medicines.
Keep VELETRI powder in a cool, dry place where it stays below 25°C.
Protect from light by keeping it in the carton until use.
Do not store VELETRI powder in a bathroom or near a sink.
Do not leave VELETRI powder in the car or on window sills.
Do not freeze VELETRI powder at any time.
Do not use this medicine after the expiry date which is stated on the carton after EXP. The expiry date refers to the last day of that month.
The reconstituted solution should be immediately further diluted to the final concentration. VELETRI diluted to the final concentration in the drug delivery reservoir as directed can be stored for up to 8 days at 2° to 8°C. Discard any unused solution after this time.
Do not freeze. Do not expose this solution to direct sunlight.
Do not use this medicine if you notice any particles in the reconstituted solution.
Product description
What it looks like
VELETRI: A Sterile, lyophilised white to off-white powder in a clear glass vial with a rubber stopper and an aluminium flip-off cap. Each pack contains one vial. The 0.5 mg vial has a white flip-off cap and the 1.5 mg vial has a red flip-off cap.
Pack size
Each pack contains one vial.
VELETRI - 0.5 mg vial of epoprostenol
VELETRI - 1.5 mg vial of epoprostenol
Ingredients
Active ingredient:
VELETRI contains the active ingredient epoprostenol, as the sodium salt.
Inactive ingredients:
- sucrose,
- L-arginine,
- sodium hydroxide (for pH adjustment)
Sponsor
JANSSEN-CILAG Pty Ltd
1-5 Khartoum Road
Macquarie Park NSW 2113 Australia
Telephone: 1800 226 334
NZ Office: Auckland New Zealand
Telephone: 0800 800 806
This leaflet was prepared in January 2024.
VELETRI 0.5 mg - AUST R 208316
VELETRI 1.5 mg - AUST R 207547
Published by MIMS March 2024
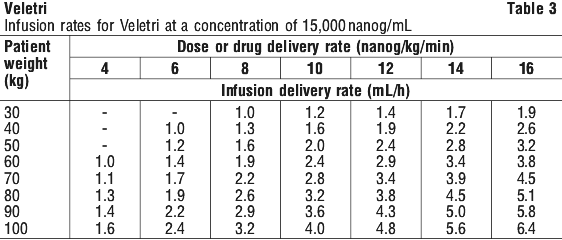
 Infusion rates may be calculated using the following formula (see Equation 1):
Infusion rates may be calculated using the following formula (see Equation 1): Examples of some concentrations commonly used in PAH are shown in Table 2 and Table 3.
Examples of some concentrations commonly used in PAH are shown in Table 2 and Table 3.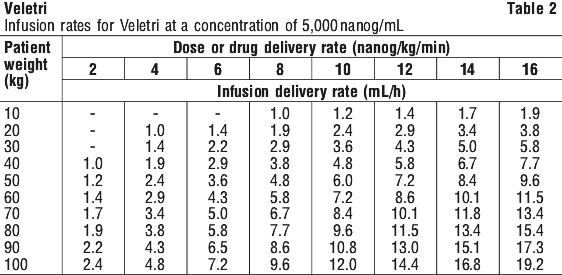
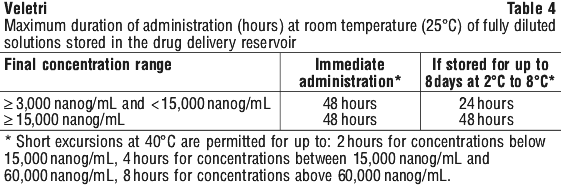
 Do not freeze. Do not expose this solution to direct sunlight.
Do not freeze. Do not expose this solution to direct sunlight.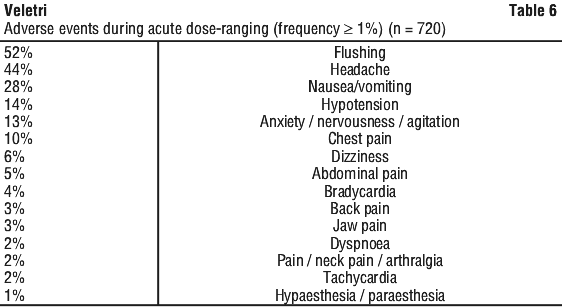 Dose limiting adverse events occurring in 1% or more of patients during acute dose-ranging were (in descending order of frequency): headache, nausea/vomiting, flushing, hypotension, anxiety/nervousness/agitation, chest pain, dizziness, bradycardia, abdominal pain, jaw pain, tachycardia, back pain, and dyspnoea.
Dose limiting adverse events occurring in 1% or more of patients during acute dose-ranging were (in descending order of frequency): headache, nausea/vomiting, flushing, hypotension, anxiety/nervousness/agitation, chest pain, dizziness, bradycardia, abdominal pain, jaw pain, tachycardia, back pain, and dyspnoea.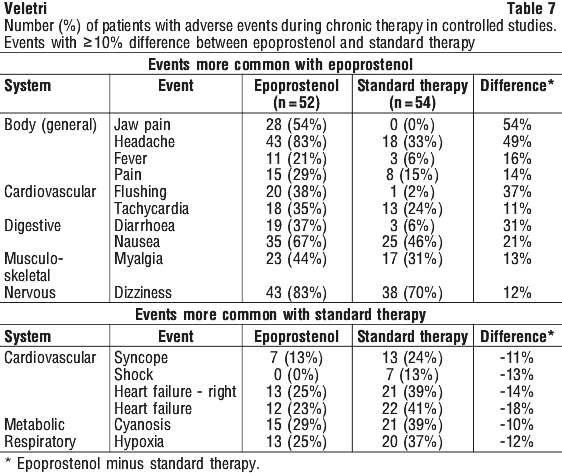 The following adverse events led to dose adjustment or discontinuation of epoprostenol in ≥ 1% of patients: dyspnoea, nausea, asthenia, flushing, headache, chest pain, diarrhoea, dizziness, vomiting, hypotension, pallor, myalgia, jaw pain, pain, and syncope.
The following adverse events led to dose adjustment or discontinuation of epoprostenol in ≥ 1% of patients: dyspnoea, nausea, asthenia, flushing, headache, chest pain, diarrhoea, dizziness, vomiting, hypotension, pallor, myalgia, jaw pain, pain, and syncope.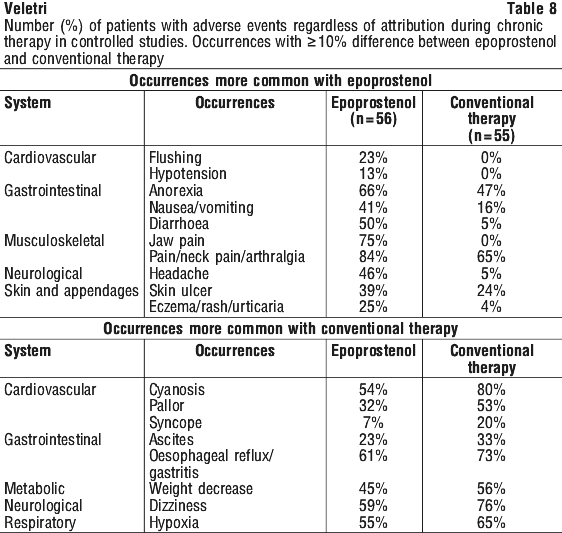
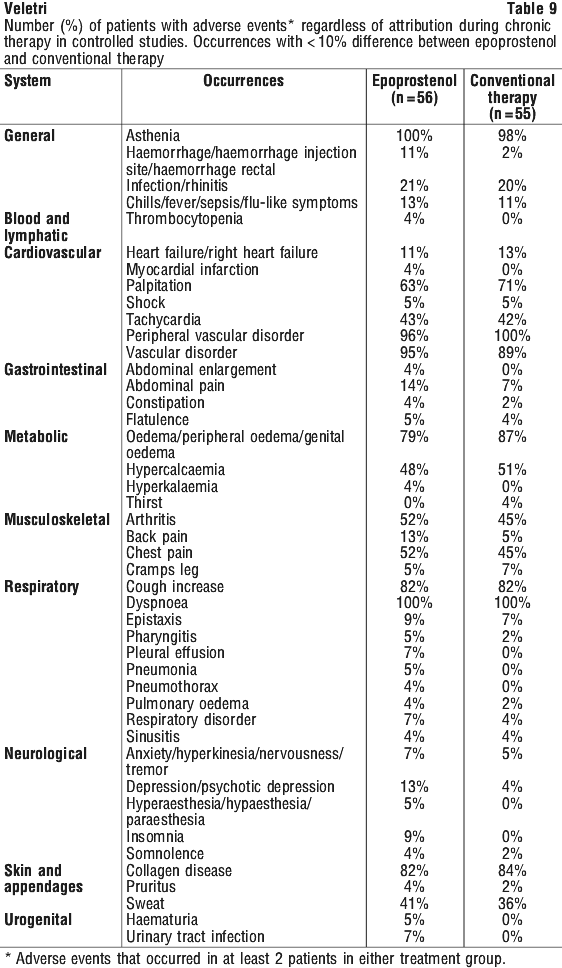
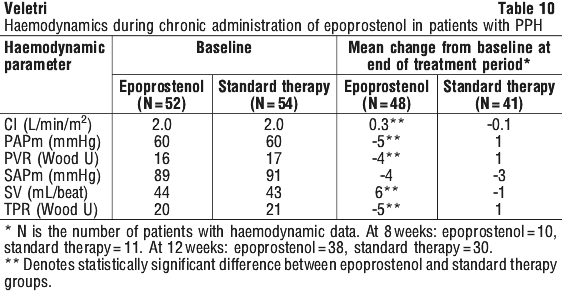 These haemodynamic improvements appeared to persist for at least 18 months when epoprostenol was administered in an open, uncontrolled study.
These haemodynamic improvements appeared to persist for at least 18 months when epoprostenol was administered in an open, uncontrolled study.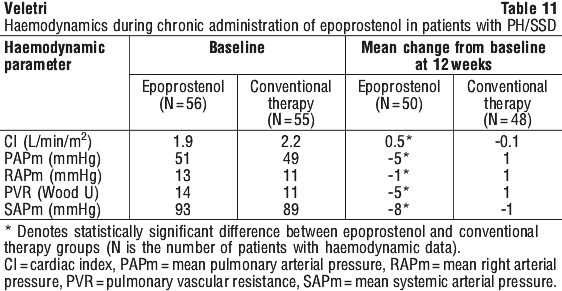
 Molecular formula: C20H31NaO5.
Molecular formula: C20H31NaO5.