1 Name of Medicine
Etrasimod.
2 Qualitative and Quantitative Composition
Each film-coated tablet contains 2.762 mg of etrasimod arginine, equivalent to 2 mg etrasimod.
Excipient(s) with known effect.
Contains tartrazine.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Film-coated tablet.
Velsipity is a green, round, film-coated tablet of approximately 6 mm diameter, debossed with "ETR" on one side and "2" on the other side.
4.1 Therapeutic Indications
Velsipity is indicated for the treatment of adults with moderately to severely active ulcerative colitis (UC) who have had inadequate response, loss of response or intolerance to conventional, biologic or Janus kinase (JAK) inhibitor therapies.
4.2 Dose and Method of Administration
Dosage.
The recommended dose of Velsipity is 2 mg taken orally once daily.
Treatment with Velsipity should be initiated under the supervision of a physician experienced in the management of conditions for which Velsipity is indicated.
Before initiating treatment with Velsipity, assess the following in Table 1.

Method of administration.
Velsipity should be swallowed whole and can be administered with or without food (see Section 5.1 Pharmacodynamic Properties; Section 5.2 Pharmacokinetic Properties).
Missed dose.
If a dose is missed, the prescribed dose should be taken at the next scheduled time; the next dose should not be doubled.
Dosage adjustments.
Paediatric population.
The safety and effectiveness of Velsipity in paediatric patients have not been established.
Elderly.
No dose adjustment is needed in patients over 65 years of age.
There are limited data available on patients over 65 years of age. No clinically significant differences in the pharmacokinetics of etrasimod were observed based on age from a population pharmacokinetics model (see Section 5.2 Pharmacokinetic Properties). In general, use of etrasimod in elderly patients should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Hepatic impairment.
No dose adjustment is needed for patients with mild or moderate hepatic impairment. Velsipity is not recommended in patients with severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
Velsipity is contraindicated in the following circumstances:
Patients who in the last 6 months, have experienced a myocardial infarction, unstable angina pectoris, stroke, transient ischemic attack (TIA), decompensated heart failure requiring hospitalisation, or New York Heart Association (NYHA) Class III/IV heart failure.
Patients with a history or presence of Mobitz type II second-degree or third-degree atrioventricular (AV) block, sick sinus syndrome, or sino-atrial block, unless the patient has a functioning pacemaker.
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
During pregnancy and in women of childbearing potential not using effective contraception.
In breastfeeding women.
Active malignancies.
4.4 Special Warnings and Precautions for Use
Bradyarrhythmia and atrioventricular conduction delays.
Prior to treatment initiation with Velsipity, an electrocardiogram (ECG) in all patients should be obtained to assess for pre-existing cardiac conduction abnormalities.
Initiation of Velsipity may result in a transient decrease in heart rate and AV conduction delays (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties).
Caution should be applied when Velsipity is initiated in patients receiving treatment with a beta‑blocker because of the potential additive effects on lowering heart rate. Similar caution should be applied if patients receive calcium channel blockers, QT prolonging medicinal products, Class Ia and Class III antiarrhythmic substances (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions), since co-administration of these substances with Velsipity may lead to additive effects. Temporary interruption of beta blocker or other antiarrhythmic treatments may be needed prior to initiation of Velsipity, depending on the resting HR before initiation of Velsipity (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). If interruption is deemed necessary, consider seeking the advice of a cardiologist.
Beta blocker or other antiarrhythmic agents treatment can be initiated in patients who have received Velsipity for at least 7 consecutive days.
On Day 1, after the first dose of Velsipity 2 mg in UC patients, the greatest mean decrease from baseline in heart rate was 7.3 bpm at Hour 3 in ELEVATE UC 52 and Hour 2 in ELEVATE UC 12. In ELEVATE UC 12, two subjects in the etrasimod group had a heart rate of less than 50 bpm, leading to withdrawal from study and recovery without medical intervention for heart rate post termination.
Patients who experienced bradycardia were generally asymptomatic. Few patients experienced symptoms, such as dizziness, and these symptoms resolved without intervention.
If treatment with Velsipity is considered, advice from a cardiologist should be sought, for those individuals:
with significant QT prolongation (QTcF ≥ 450 msec in males, ≥ 470 msec in females);
with arrhythmias requiring treatment with Class Ia or Class III antiarrhythmic drugs;
with unstable ischemic heart disease, heart failure, history of cardiac arrest, cerebrovascular disease (occurring more than 6 months prior to treatment initiations), or uncontrolled hypertension;
with resting heart rate of less than 50 bpm;
with history of symptomatic bradycardia, recurrent cardiogenic syncope, or severe untreated sleep apnea;
with history of Mobitz type I second-degree AV block, unless the patient has a functioning pacemaker.
First dose monitoring in patients with certain pre-existing cardiac conditions.
Due to risk of transient decreases in heart rate with the initiation of etrasimod 4-hour monitoring of signs and symptoms of symptomatic bradycardia after the first dose is recommended in patients with resting heart rate < 50 bpm, second-degree [Mobitz type I] AV block, or a history of myocardial infarction or heart failure.
Patients should be monitored with hourly pulse and blood pressure measurement during this 4-hour period. An ECG prior to and at the end of this 4-hour period is recommended.
Additional monitoring is recommended in patients, if at the end of 4-hour period:
Heart rate is < 45 bpm.
Heart rate is the lowest value post dose, suggesting that the maximum decrease in heart rate may not have occurred yet.
ECG shows evidence of a new onset second-degree or higher AV block.
Infections.
Risk of infections.
Velsipity causes a mean reduction in peripheral blood lymphocyte count to approximately 45% of baseline values at Week 52 because of reversible sequestration of lymphocytes in lymphoid tissues (see Section 5.1 Pharmacodynamic Properties). Velsipity may, therefore, increase the susceptibility to infections (see Section 4.8 Adverse Effects (Undesirable Effects)).
Before initiating treatment, obtain a recent complete blood count (CBC), including lymphocyte count (i.e. within the last 6 months or after discontinuation of prior UC therapy). Clinicians should monitor complete blood count (CBC) periodically during treatment and to interrupt treatment with etrasimod in patients with a confirmed absolute lymphocyte count < 0.2 x 109/L until the level reaches > 0.5 x 109/L when re-initiation of etrasimod can be considered. Patients need to report symptoms of infection promptly to their physician.
The initiation of Velsipity in patients with any active infection should be delayed until the infection is resolved.
Consider interruption of treatment with Velsipity if a patient develops a serious infection.
Because residual pharmacodynamic effects, such as lowering effects on peripheral lymphocyte count, may persist up to 2 weeks after discontinuation of Velsipity, vigilance for infection should be continued throughout this period (see Section 5.1 Pharmacodynamic Properties).
Progressive multifocal leukoencephalopathy.
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the brain caused by the John Cunningham virus (JCV) that typically occurs in patients who are immunocompromised, and that may lead to death or severe disability. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
No cases of PML have been reported in Velsipity-treated patients in the development program; however, PML has been reported in multiple sclerosis patients treated with other sphingosine 1-phosphate (S1P) receptor modulators and has been associated with some risk factors (e.g. immunocompromised patients, polytherapy with immunosuppressants). Physicians should be vigilant for clinical symptoms or unexplained neurologic findings that may be suggestive of PML. If PML is suspected, treatment with Velsipity should be suspended until PML has been excluded by an appropriate diagnostic evaluation.
If PML is confirmed, treatment with Velsipity should be discontinued.
Prior and concomitant treatment with antineoplastic, immune-modulating, or immunosuppressive therapies.
In ELEVATE UC 52 and ELEVATE UC 12, patients who received Velsipity were not to receive concomitant treatment with antineoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies used for the treatment of UC. In ELEVATE UC 52 and ELEVATE UC 12, concomitant use of corticosteroids was allowed; however long-term data on concomitant use of etrasimod and corticosteroids are limited and did not appear to influence the safety or efficacy of Velsipity (see Section 5.1 Pharmacodynamic Properties).
Caution should be used when co-administering etrasimod and antineoplastic, immune-modulating, or immunosuppressive (including corticosteroid) therapies to patients, because of the risk of additive immune system effects during such therapy (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
When switching to Velsipity from immunosuppressive medications, consider the duration of their effects and their mode of action to avoid unintended additive immune system effects.
An appropriate washout period may need to be applied.
After stopping Velsipity, lymphocyte counts returned to the normal range in 90% of subjects within 1 to 2 weeks of stopping Velsipity based on a population pharmacokinetic/pharmacodynamic model (see Section 5.1 Pharmacodynamic Properties). Use of immunosuppressants within this period may lead to an additive effect on the immune system, and therefore monitor patients receiving concomitant immunosuppressants for infectious complications up to 2 weeks after the last dose of Velsipity.
Vaccinations.
No clinical data are available on the safety and efficacy of vaccinations in patients taking Velsipity. Vaccinations may be less effective if administered during Velsipity treatment. If live attenuated vaccine immunisations are required, administer at least 4 weeks prior to initiation of Velsipity. Avoid the use of live attenuated vaccines during and for at least 2 weeks after treatment with Velsipity.
Patients without a healthcare professional-confirmed history of varicella (chickenpox) or without documentation of a full course of vaccination against varicella zoster virus (VZV) should be tested for antibodies to VZV before initiating Velsipity. A full course of VZV vaccination of antibody-negative patients is recommended prior to commencing treatment with Velsipity, following which initiation of treatment with Velsipity should be postponed for 4 weeks to allow the full effect of vaccination to occur.
Update immunisations in agreement with current immunisation guidelines prior to initiating Velsipity therapy.
Increased blood pressure.
In clinical studies, hypertension was more frequently reported in patients treated with Velsipity than in patients treated with placebo (see Section 4.8 Adverse Effects (Undesirable Effects)). Blood pressure should be monitored during treatment with Velsipity and managed appropriately.
Fetal risk.
Based on animal studies, Velsipity may cause fetal harm (see Section 4.6 Fertility, Pregnancy and Lactation). Before initiation of Velsipity treatment, women of childbearing potential should be counseled on the potential for a serious risk to the fetus and the need for effective contraception during treatment with Velsipity.
Because it takes approximately 7 days for pharmacokinetic/pharmacodynamic effects of Velsipity to be washed out from the body, women of childbearing potential should use effective contraception to avoid pregnancy during treatment and for 7 days after stopping Velsipity (see Section 4.6 Fertility, Pregnancy and Lactation).
Macular oedema.
S1P receptor modulators, including Velsipity, have been associated with an increased risk of macular oedema. Macular oedema with or without visual symptoms has been reported in 0.3% of patients in ELEVATE UC 12 and ELEVATE UC 52 trials treated with Velsipity.
Patients with a history of diabetes mellitus, uveitis, or underlying/co-existing retinal disease, uncontrolled hypertension and older patients are at increased risk of macular oedema during Velsipity therapy (see Section 4.8 Adverse Effects (Undesirable Effects)). Arrange an ophthalmological assessment in these patients prior to treatment initiation with Velsipity and have follow up evaluations while receiving therapy.
In patients without the risk factors above, an ophthalmic evaluation of the fundus, including the macula, is recommended within 3-4 months after starting Velsipity treatment (cases reported with Velsipity occurred within this timeframe) and at any time if there is a change in vision while taking Velsipity.
Patients who present with visual symptoms of macular oedema should be evaluated and, if confirmed, treatment with Velsipity should be discontinued. A decision on whether or not Velsipity should be re-initiated after resolution needs to take into account the potential benefits and risks for the individual patient.
Malignancies.
Cases of malignancies (including skin malignancies) have been reported in patients treated with S1P receptor modulators. If a suspicious skin lesion is observed, it should be promptly evaluated.
Since there is a potential risk of malignant skin growths, patients treated with Velsipity should have periodic skin examinations and be cautioned against exposure to sunlight without protection. These patients should not receive concomitant phototherapy with UV-B radiation or PUVA photochemotherapy.
Posterior reversible encephalopathy syndrome.
Rare cases of posterior reversible encephalopathy syndrome (PRES) have been reported in patients receiving other S1P receptor modulators. Such events have not been reported for Velsipity-treated patients in the development program. Should a Velsipity-treated patient develop any neurological or psychiatric symptoms/signs (e.g. cognitive deficits, behavioural changes, cortical visual disturbances, or any other neurological cortical symptoms/signs), any symptom/sign suggestive of an increase of intracranial pressure, or accelerated neurological deterioration, the physician should promptly schedule a complete physical and neurological examination and should consider an MRI. Symptoms of PRES are usually reversible but may evolve into ischemic stroke or cerebral hemorrhage. Delay in diagnosis and treatment may lead to permanent neurological sequelae. If PRES is suspected, treatment with Velsipity should be discontinued.
Respiratory effects.
Reductions in absolute forced expiratory volume over 1 second (FEV1) and forced vital capacity (FVC) were observed in patients treated with S1P receptor modulators, including Velsipity (see Section 5.1 Pharmacodynamic Properties). Patients with a forced expiratory volume in 1 second (FEV1) or forced vital capacity (FVC) < 70% predicted values and FEV1/FVC ratio < 0.70 at screening were not eligible to participate in ELEVATE 12 and ELEVATE 52. Velsipity should be used with caution in patients with severe respiratory disease (i.e. pulmonary fibrosis, asthma, and chronic obstructive pulmonary disease).
Use in hepatic impairment.
Elevations of aminotransferases may occur in patients receiving Velsipity (see Section 4.8 Adverse Effects (Undesirable Effects)).
Recent (i.e. within last 6 months) transaminase and bilirubin levels should be available before initiation of treatment with Velsipity.
Patients who develop symptoms suggestive of hepatic dysfunction, such as unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine, should have hepatic enzymes checked and Velsipity should be discontinued if significant liver injury is confirmed.
Velsipity is not recommended in severe hepatic impairment. See Section 5.2 Pharmacokinetic Properties.
Use in the elderly.
See Section 4.2 Dose and Method of Administration, Dosage adjustments, Elderly; Section 5.2 Pharmacokinetic Properties.
Paediatric use.
The safety and effectiveness of Velsipity in paediatric patients have not been established.
Effects on laboratory tests.
See Section 4.4 Special Warnings and Precautions for Use, Infections, Risk of infections and Use in hepatic impairment; Section 4.8 Adverse Effects (Undesirable Effects), Description of selected adverse reactions, Blood lymphocyte count reduction and Elevated hepatic enzymes; Section 5.1 Pharmacodynamic Properties, Pharmacodynamic effects, Reduction in blood lymphocyte counts and Reduction in tissue lymphocyte counts.4.5 Interactions with Other Medicines and Other Forms of Interactions
Effect of other medicinal products on etrasimod.
In vitro studies indicate that metabolism of etrasimod occurs through multiple distinct enzyme systems, including multiple CYP450 (CYP2C8, CYP2C9, and CYP3A4), non-CYP450 oxidative enzymes and UGTs. Metabolism by sulfotransferases was observed in clinical excreta samples based on metabolite profiling. Overall, the disposition of etrasimod is mediated by several enzymes without major contribution by any single enzyme.
Etrasimod is not a substrate of P-gp, BCRP, OATP1B1/3, OAT1/3, OCT1/2 transporters. Drugs that are inhibitors of these transporters are unlikely to impact the pharmacokinetics of etrasimod.
CYP2C8, CYP2C9 and CYP3A4 inhibitors.
The co-administration of etrasimod with steady state fluconazole (moderate CYP2C9 and CYP3A4 inhibitor) increased exposure (AUC) of etrasimod by 84%. Co-administration of etrasimod with a therapeutic agent or a combination of agents that are moderate to strong inhibitors of two or more of the following CYPs (CYP2C8, CYP2C9, and CYP3A4) (e.g. fluconazole) increases the exposure of etrasimod and is not recommended.
Due to the potential for increased exposure of etrasimod, co-administration of etrasimod in patients who are known or suspected to be CYP2C9 poor metabolisers and who take medicinal products that are moderate or strong inhibitors of CYP2C8 or CYP3A4 is not recommended.
CYP2C8, CYP2C9, and CYP3A4 inducers.
The co-administration of etrasimod with rifampicin (strong CYP3A4, moderate CYP2C8, and CYP2C9 inducer) decreased exposure (AUC) of etrasimod by 49%. Co-administration of Velsipity with a therapeutic agent or a combination of agents that are moderate to strong inducers of two or more of the main metabolizing CYPs (CYP2C8, CYP2C9, and CYP3A4) decreases the exposure of etrasimod. Co-administration with Velsipity and such agents (e.g. rifampicin) is not recommended.
Concomitant drugs that may decrease heart rate.
Beta blockers and calcium channel blockers.
The co-administration of Velsipity in patients receiving stable beta blocker treatment did not result in additive effects on heart rate reduction. Velsipity can be initiated in patients receiving stable doses of beta blocker treatment. Following the first dose of etrasimod 2 mg, the Day 1 maximum mean change from baseline heart rate reduction in patients on stable beta blocker treatment was comparable to patients not taking a beta blocker (mean [SD]: -6.5 [7.15] bpm compared with -7.2 [9.27] bpm).
The initiation of a beta blocker in patients receiving stable treatment of Velsipity has not been studied.
The effect of co-administration of Velsipity and a calcium channel blocker has not been studied.
Caution is recommended for patients receiving medicinal products that slow heart rate or atrioventricular conduction because of the potential additive effects on lowering heart rate (see Section 4.4 Special Warnings and Precautions for Use).
Antiarrhythmic drugs and QT prolonging drugs.
Velsipity has not been studied in patients taking QT prolonging drugs.
Class Ia (e.g. quinidine, procainamide) and Class III (e.g. amiodarone, sotalol) antiarrhythmic drugs have been associated with cases of Torsades de Pointes in patients with bradycardia. If treatment with Velsipity is considered in patients on Class Ia or Class III antiarrhythmic drugs, advice from a cardiologist should be sought (see Section 4.4 Special Warnings and Precautions for Use).
Because of the potential additive effects on heart rate, if treatment initiation with Velsipity is considered in patients on QT prolonging drugs, advice from a cardiologist should be sought (see Section 4.4 Special Warnings and Precautions for Use).
Antineoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies.
Velsipity has not been studied in combination with antineoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies. Caution should be used during concomitant administration because of the risk of additive immune system effects during such therapy and in the weeks following administration (see Section 4.4 Special Warnings and Precautions for Use).
Vaccination.
Vaccinations may be less effective if administered during and for up to 2 weeks after discontinuation of Velsipity treatment. The use of live attenuated vaccine may carry the risk of infection and should therefore be avoided during Velsipity treatment and for at least 2 weeks after discontinuation of Velsipity treatment (see Section 4.4 Special Warnings and Precautions for Use).
Effect of etrasimod on other drugs.
In vitro studies indicate that at the recommended dose of 2 mg once daily, etrasimod is unlikely to show any clinically relevant drug-drug interaction potential for CYP450 or UGT enzymes or membrane transporters.
Oral contraceptives.
No clinically significant differences in the pharmacokinetics and pharmacodynamics of oral contraceptive containing 30 microgram ethinylestradiol and 150 microgram levonorgestrel were observed when co-administered with etrasimod.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
The effect of etrasimod on human fertility has not been evaluated.
When etrasimod was administered orally to male (up to 200 mg/kg/day) and female (up to 4 mg/kg/day) rats daily from pre-mating to conception and conception to implantation, there were no adverse effects observed on male or female fertility. Estimated plasma etrasimod exposure (AUC) at the highest dose tested was approximately 750 (males) and 22 (females) times that in humans at the maximum recommended human dose (MRHD) or 2 mg.
(Category D)
There are no adequate and well-controlled studies on the developmental risk associated with the use of Velsipity in pregnant women. Velsipity should not be used during pregnancy (see Section 4.3 Contraindications).
In animal studies, administration of etrasimod during pregnancy produced adverse effects on development, including embryolethality and fetal malformations, in both rats and rabbits at clinically relevant maternal exposures.
When etrasimod was orally administered to pregnant rats during the period of organogenesis, post-implantation loss with a corresponding lower mean number of viable fetuses was observed at 4 mg/kg/day. Etrasimod-related fetal external malformations of anasarca and localised oedema (at 4 mg/kg/day) and visceral malformations (including aortic vessel anomalies and septal defects) and variations (short brachiocephalic trunk) at ≥ 1 mg/kg/day were noted. Maternal plasma AUC at the lowest dose tested (1 mg/kg/day), which was a teratogenic dose, was 6 times that in humans at the MRHD of 2 mg/day.
When etrasimod was orally administered to pregnant rabbits during the period of organogenesis, post-implantation loss with a corresponding lower number of viable fetuses was observed at ≥ 10 mg/kg/day. Etrasimod-related fetal visceral malformations of the aortic arch (bulbous aortic arch, retroesophageal aortic arch and/or coarction of the aortic arch) were observed at ≥ 10 mg/kg/day and fetal skeletal malformations (fused sternebrae) at 20 mg/kg/day. The fetal skeletal variation of extra ossification site anterior to the sternebra was also noted at ≥ 10 mg/kg/day. There were no effects on embryofetal development at 2 mg/kg/day. Maternal plasma exposure (AUC) at the no-adverse-effect dose (2 mg/kg/day) was 0.9 times that in humans at the MRHD of 2 mg/day.
Oral administration of etrasimod to female rats throughout pregnancy and lactation resulted in prolonged gestation, increased post-implantation loss and an increased number of stillbirths at ≥ 2 mg/kg/day. Maternal plasma exposure (AUC) at the no observed adverse effect level (0.4 mg/kg/day) was 1.2 times that in humans at the MRHD of 2 mg/day.
Women of childbearing potential.
Before initiation of Velsipity treatment, women of childbearing potential should be counseled on the potential for a serious risk to the fetus and the need for effective contraception during treatment with Velsipity.
Because of the time it takes to eliminate the drug from the body after stopping treatment, the potential risk to the fetus may persist and women of childbearing potential should use effective contraception for 7 days after stopping Velsipity (see Section 4.3 Contraindications).
It is unknown whether Velsipity is excreted in human milk. When etrasimod was orally administered to female rats during pregnancy and lactation, etrasimod was detected in the plasma of the offspring, suggesting excretion of etrasimod in milk. A risk to newborns/infants cannot be excluded. Women receiving Velsipity should not breastfeed.
Oral administration of etrasimod (0, 0.4, 2, or 4 mg/kg/day) to female rats throughout pregnancy and lactation resulted in decreased mean pup weights at all dose levels during the preweaning period, lower pup viability at 2 and 4 mg/kg/day, and reduced fertility and reproductive performance (reduction in implantations and increased preimplantation loss) in F1 pups at the highest dose tested. No effects were noted on neurobehavioural function in offspring at any dose level tested. Plasma exposure (AUC) in dams at the lowest dose tested was equivalent (1.2 times) to those in humans at the MRHD of 2 mg/day.4.7 Effects on Ability to Drive and Use Machines
Velsipity has no or negligible influence on patient's ability to drive and use machines. However, when driving or using machines, it should be taken into account that dizziness has been reported (see Section 4.8 Adverse Effects (Undesirable Effects)). No studies on the effects on the ability to drive and the use of machines have been performed.
4.8 Adverse Effects (Undesirable Effects)
Summary of safety profile.
The most common adverse drug reactions are lymphopenia (11%) and headache (7%).
Tabulated list of adverse reactions.
The adverse reactions observed in patients treated with etrasimod are listed by system organ class (SOC) and frequency for all adverse reactions. Within each SOC and frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000). See Table 2.
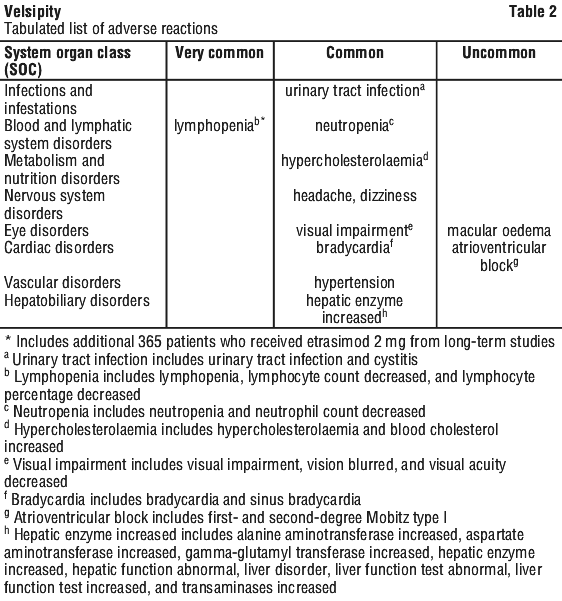
Description of selected adverse reactions.
Bradyarrhythmia.
In ELEVATE UC 52, bradycardia was reported on the day of treatment initiation in 1.0% of patients treated with Velsipity compared to none in patients who received placebo. On Day 2, bradycardia was reported in 1 patient (0.3%) treated with Velsipity compared to none in patients who received placebo. In ELEVATE UC 12, bradycardia was reported on the day of treatment initiation in 2.1% of patients treated with Velsipity compared to none in patients who received placebo. On Day 2, bradycardia was reported in 1 patient (0.4%) treated with Velsipity compared to none in patients who received placebo.
At initiation of Velsipity 2 mg, events of first- or second-degree Mobitz type I AV blocks were observed in 0.7% of Velsipity-treated patients compared to none in placebo in ELEVATE UC 52 and in 0.4% of Velsipity-treated patients compared to none in placebo in ELEVATE UC 12; however, in ELEVATE UC 52 and ELEVATE UC 12, Mobitz type II second- or third-degree AV blocks were not reported in patients treated with Velsipity.
Infections.
In ELEVATE UC 52, the overall rate of infections and rate of serious infections in patients treated with Velsipity was comparable to that in patients who received placebo (24.9% vs. 22.2%, and 1.0% vs. 3.5%, respectively). In ELEVATE UC 12, the overall rate of infections and rate of serious infections in patients treated with Velsipity was comparable to that in patients who received placebo (11.3% vs. 12.1%, and none in both groups, respectively). The most common adverse reaction for infections was urinary tract infection.
Blood lymphocyte count reduction.
The proportion of patients treated with Velsipity who experienced lymphocyte counts less than 0.2 x 109/L was 5.6% in ELEVATE UC 52 and 0.9% in ELEVATE UC 12. These events did not lead to treatment discontinuation.
Elevated hepatic enzymes.
In ELEVATE UC 52, elevations of alanine aminotransferase (ALT) to 5-fold the upper limit of normal (ULN) or greater occurred in 0.7% of patients treated with Velsipity and 0.7% of patients who received placebo, and in ELEVATE UC 12 elevations occurred in 0.8% of patients treated with Velsipity and no patients who received placebo. In ELEVATE UC 52, elevations of ALT to 3-fold the ULN or greater occurred in 4.5% of patients treated with Velsipity and 0.7% of patients who received placebo, and in ELEVATE UC 12 elevations occurred in 2.5% of patients treated with Velsipity and no patients who received placebo.
The majority (75%) of patients with ALT greater than 3-fold the ULN continued treatment with Velsipity with values returning to less than 3-fold the ULN while on treatment.
Overall, the percentage of discontinuation because of elevations in hepatic enzymes was 0.4% in patients treated with Velsipity, and 0.4% in patients who received placebo.
Increased blood pressure.
In ELEVATE UC 52 and ELEVATE UC 12, patients treated with Velsipity had an average increase of approximately 1 to 4 mmHg in systolic blood pressure and approximately 1 to 2 mmHg in diastolic blood pressure compared to < 1.5 mmHg and < 1 mmHg in patients receiving placebo, respectively. The increase was first detected after 2 weeks of treatment and remained within the specified average range in blood pressure increases throughout treatment. Hypertension was reported as an adverse reaction in 2.1% of patients treated with Velsipity and in 1.0% of patients who received placebo. The majority of the events were mild to moderate in severity.
Macular oedema.
In ELEVATE UC 52, macular oedema was reported in 0.3% of patients treated with Velsipity and in no patients receiving placebo. In ELEVATE UC 12, macular oedema was reported in 0.4% of patients treated with Velsipity and in 0.9% of patients receiving placebo.
Herpes viral infections.
Cases of localised herpes viral infection were seen with S1P receptor modulators, including Velsipity. In ELEVATE UC 52, herpes zoster was reported in 0.7% of patients treated with Velsipity and in none of the patients who received placebo. In ELEVATE UC 12, herpes zoster was reported in none of the patients treated with Velsipity and in 1.7% of patients who received placebo.
Tabulated list of adverse events.
Treatment-emergent adverse events (TEAE) reported by ≥ 2% of subjects in any treatment group for the 12-week induction period (pool includes APD334-301, APD334-302 studies) and 40-week maintenance period (APD334-301) are provided in Table 3 and Table 4, respectively.


Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/safety/reporting-problems.4.9 Overdose
In patients with overdosage of etrasimod, monitor for signs and symptoms of bradycardia, which may include overnight monitoring. Regular measurements of heart rate, blood pressure, and ECGs should be performed. There is no specific antidote to etrasimod available.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Etrasimod is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1, 4 and 5 (S1P1,4,5). Etrasimod has no activity on S1P2 or S1P3. Etrasimod partially and reversibly blocks the capacity of lymphocytes to egress from lymphoid organs, reducing the number of lymphocytes in peripheral blood thereby lowering the number of activated lymphocytes in the tissue.
The mechanism by which etrasimod exerts therapeutic effects in UC is unknown but may involve the reduction of lymphocyte migration into the intestines.
Pharmacodynamic effects.
Heart rate and rhythm.
Velsipity may result in a transient decrease in heart rate and AV conduction upon treatment initiation (see Section 4.4 Special Warnings and Precautions for Use). On Day 1, in UC patients from ELEVATE UC 52 and ELEVATE UC 12, the greatest mean decrease in heart rate was observed at Hour 2 or 3 post dose. On day 1, the mean (SD) change in PR interval from predose to 4 hours post dose with etrasimod was 5.5 msec (18.84). PR interval prolongation > 200 msec was recorded on ECG in 5.1% and higher degree prolongation (> 230 msec) in 1.8% of subjects. The first dose heart rate lowering effect of etrasimod over the first few hours post dose may be partially attenuated following administration with food (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Effect on QT interval.
In a thorough QT study, daily administration of etrasimod doses 2 mg (recommended dose) to 4 mg (two times recommended dose) were evaluated in healthy subjects. Etrasimod did not prolong QTc interval to any clinically relevant extent.
Reduction in blood lymphocyte counts.
In controlled clinical studies, mean lymphocyte counts decreased to approximately 50% of baseline at 2 weeks (approximate mean blood lymphocyte counts 0.9 x 109/L) consistent with the mechanism of action, and lowered lymphocyte counts were maintained during once daily treatment with Velsipity.
Peripheral blood B cells [CD19+] and T cells [CD3+], T-helper [CD3+CD4+], and T-cytotoxic [CD3+CD8+] cell subsets were all reduced, while natural killer cells and monocytes were not. T-helper cells were more sensitive to the effects of etrasimod than T-cytotoxic cells.
Peripheral blood absolute lymphocyte counts returned to the normal range in 90% of patients within 1 to 2 weeks of stopping therapy based on a population pharmacokinetic/pharmacodynamic model.
Reduction in tissue lymphocyte counts.
In ELEVATE UC 52 and ELEVATE UC 12, etrasimod reduced activated lymphocytes in colon biopsies from patients with UC.
Peripheral inflammatory proteins.
Etrasimod reduces peripheral inflammatory proteins including those related to UC.
Pulmonary function.
Reductions in FEV1 and FVC were observed in patients treated with Velsipity. In ELEVATE UC 52 and ELEVATE UC 12, by Week 12, the absolute change in mean FEV1 in patients treated with Velsipity was -49 mL, compared to -19 mL for placebo. There was no further decline relative to placebo by Week 52. By Week 12 the absolute change in mean FVC in patients treated with Velsipity was -12 mL, compared to -5 mL for placebo, and at Week 52 it was -39 mL vs. 8 mL. The absolute change in mean FEV1/FVC in patients treated with Velsipity was 0.026, compared to 0.024 for placebo. There was no further decline relative to placebo by Week 52.
Clinical trials.
The efficacy of etrasimod was evaluated in 2 randomised, double-blind, placebo-controlled clinical studies (ELEVATE UC 52 and ELEVATE UC 12) in patients 16 to 80 years of age with moderately to severely active ulcerative colitis.
Both studies included patients who had an inadequate response, loss of response, or intolerance to one or more of the following treatment options: oral aminosalicylates, corticosteroids, thiopurines, Janus kinase (JAK) inhibitors, or a biologic (e.g. TNF blocker, anti-integrin, anti-IL12/23). Enrolled patients had UC confirmed by endoscopy and histopathology with the extent of disease being ≥ 10 cm from the anal verge. Patients with isolated proctitis were also included in the study provided they met all other inclusion criteria.
Disease severity was assessed on the modified Mayo score (mMS), a 3-component Mayo score (0 to 9) which consists of the following subscores (0 to 3 for each subscore): stool frequency (SF), rectal bleeding (RB), and findings on centrally read endoscopy score (ES). An ES of 2 was defined by marked erythaema, lack of vascular pattern, any friability, and/or erosions, and a score of 3 was defined by spontaneous bleeding and ulceration. Enrolled patients had a mMS of 4 to 9 with an ES ≥ 2 and RB subscore ≥ 1.
Patients in these studies may have received other concomitant UC therapies including stable daily doses of oral aminosalicylates and/or oral corticosteroids (≤ 20 mg prednisone, ≤ 9 mg budesonide, or equivalent steroid). Concomitant treatment with immunomodulators, biologic therapies, rectal 5 ASA, or rectal corticosteroids was not permitted.
ELEVATE UC 52. ELEVATE UC 52 was a treat-through study, with a total of 433 patients randomised to receive etrasimod 2 mg or placebo at a 2:1 ratio administered orally once daily. Patients remained on their assigned treatment for the duration of the study.
At baseline, enrolled patients had a median mMS of 7, with 5.5% of patients having mMS of 4, 66.5% having mMS 5 to 7 (moderately active disease), and 28% having mMS > 7 (severely active disease). 8% of enrolled patients presented with isolated proctitis. A total of 30% of patients had prior exposure to biologic/JAK inhibitors; a total of 14% of patients had exposure to > 1 biologic/JAK inhibitor and 11% of patients had prior exposure to anti-integrins. At baseline, 77% of patients were receiving oral aminosalicylates and 31% of patients were receiving oral corticosteroids.
The co-primary endpoints were the proportion of patients achieving clinical remission at Week 12 and at Week 52, with clinical remission defined as SF subscore of 0 (or 1 with a ≥ 1-point decrease from baseline), RB subscore of 0, and ES ≤ 1 (excluding friability). The secondary endpoints included the proportion of patients achieving endoscopic improvement, symptomatic remission, mucosal healing, clinical response, corticosteroid-free clinical remission, and sustained clinical remission. The primary analysis was conducted at Week 12 and at Week 52 in patients with moderately to severely active disease, defined as mMS 5 to 9 (see Table 5).
A significantly greater proportion of patients treated with etrasimod achieved clinical remission, endoscopic improvement, symptomatic remission, and mucosal healing at Week 12 and at Week 52, corticosteroid-free clinical remission and sustained clinical remission at Week 52, compared to placebo (see Table 5).

Supplementary analysis of mMS 4 to 9.
Consistent with the primary analysis (mMS of 5 to 9 including ES ≥ 2 and RB subscore ≥ 1), a greater proportion of patients with mMS of 4 to 9 (including ES ≥ 2 and RB subscore ≥ 1) treated with etrasimod compared to placebo achieved clinical remission (28% vs. 8%, 2-sided p-value < 0.001), endoscopic improvement (37% vs 17%, 2-sided p-value < 0.001), symptomatic remission (46% vs. 22%, 2-sided p-value < 0.001), and mucosal healing (23% vs. 6%, 2-sided p-value < 0.001) at Week 12, clinical remission (33% vs. 8%, 2-sided p-value < 0.001), endoscopic improvement (39% vs. 13%, 2-sided p-value < 0.001), symptomatic remission (44% vs. 19%, 2-sided p-value < 0.001), mucosal healing (27% vs. 10%, 2-sided p-value < 0.001), corticosteroid-free clinical remission (33% vs. 7%, 2-sided p-value < 0.001), and sustained clinical remission (19% vs. 3%, 2-sided p-value < 0.001) at Week 52.
Isolated proctitis.
A greater proportion of patients with isolated proctitis at baseline treated with etrasimod compared to placebo achieved clinical remission at Week 12 (46% vs. 29%) and Week 52 (42% vs. 14%).
Symptomatic remission by week 2.
At Week 2 (first study visit), a greater proportion of patients treated with etrasimod compared to placebo achieved symptomatic remission (16% vs. 11%).
Complete symptomatic remission.
Complete symptomatic remission was defined as a SF subscore of 0 and RB subscore of 0. At Week 4, a greater proportion of patients treated with etrasimod compared to placebo achieved complete symptomatic remission (11% vs. 4%).
Stool frequency and rectal bleeding subscores.
Decreases in SF and RB subscores were observed as early as Week 2 in patients treated with etrasimod compared to placebo.
Cessation of rectal bleeding.
A greater proportion of patients achieved an RB subscore of 0 as early as Week 4 with etrasimod compared to placebo (44% vs. 27%).
Endoscopic and histologic assessment.
Normalisation of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. A greater proportion of patients treated with etrasimod compared to placebo achieved endoscopic remission by Week 12 (15% vs. 4%), Week 52 (26% vs. 6%), and both Week 12 and Week 52 (11% vs. 2%).
Endoscopic remission and Geboes histologic score < 2.0 (indicating no neutrophils in crypts or lamina propria and no increase in eosinophil, no crypt destruction, and no erosions, ulcerations, or granulation tissue) were achieved by a greater proportion of patients treated with etrasimod compared to placebo at Week 12 (11% vs. 2%) and at Week 52 (18% vs. 5%).
When defined as ES ≤ 1 and Geboes ≤ 3.1 (indicating neutrophil infiltration in < 5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue), a greater proportion of patients treated with etrasimod compared to placebo achieved histologic-endoscopic mucosal improvement at Week 12 (31% vs. 10%) and Week 52 (40% vs. 11%).
Abdominal pain and bowel urgency.
At Week 12, a greater proportion of patients treated with etrasimod compared to placebo had absence of abdominal pain (27% vs. 13%) and absence of bowel urgency (19% vs. 7%). At Week 52, a greater proportion of patients treated with etrasimod compared to placebo had absence of abdominal pain (22% vs. 7%) and absence of bowel urgency (19% vs. 8%).
Inflammatory bowel disease questionnaire (IBDQ).
Patients treated with etrasimod compared to placebo demonstrated greater improvement from baseline in the total and all 4 domain scores of the IBDQ (bowel symptoms, systemic function, emotional function, and social function) at Week 12 and at Week 52.
ELEVATE UC 12. In ELEVATE UC 12, a total of 354 patients were randomised to receive etrasimod 2 mg or placebo at a 2:1 ratio administered orally once daily.
At baseline, enrolled patients had a median mMS of 7, with 5.6% of patients having mMS of 4, and 67% having mMS 5 to 7 (moderately active disease), and 27.4% having mMS > 7 (severely active disease). 8% of enrolled patients presented with isolated proctitis. A total of 33% of patients had prior exposure to biologic/JAK inhibitors; a total of 18% of patients had exposure to > 1 biologic/JAK inhibitor and 12% of patients had prior exposure to anti-integrins. At baseline, 83% of patients were receiving oral aminosalicylates and 28% of patients were receiving oral corticosteroids.
The primary endpoint was the proportion of patients achieving clinical remission at Week 12. The secondary endpoints included the proportion of patients achieving endoscopic improvement, symptomatic remission, mucosal healing, and clinical response at Week 12.
The primary analysis was conducted at Week 12 in patients with moderately to severely active disease, defined as mMS 5 to 9 (see Table 6).
A significantly greater proportion of patients treated with Velsipity achieved clinical remission, endoscopic improvement, symptomatic remission, and mucosal healing compared to placebo at Week 12 (see Table 6).

Supplementary analysis of mMS 4 to 9.
Consistent with the primary analysis (mMS of 5 to 9 including ES ≥ 2 and RB subscore ≥ 1), a greater proportion of patients with mMS of 4 to 9 (including ES ≥ 2 and RB subscore ≥ 1) treated with Velsipity compared to placebo achieved clinical remission (26% vs. 15%, 2-sided p-value = 0.007), endoscopic improvement (33% vs. 19%, 2-sided p-value = 0.002), symptomatic remission (48% vs. 29%, 2-sided p-value < 0.001), and mucosal healing (17% vs. 9%, 2-sided p-value = 0.012) at Week 12.
Isolated proctitis.
A greater proportion of patients with isolated proctitis at baseline treated with Velsipity compared to placebo achieved clinical remission at Week 12 (39% vs. 8%).
Symptomatic remission by week 4.
At Week 4, a greater proportion of patients treated with Velsipity compared to placebo achieved symptomatic remission (28% vs. 16%).
Complete symptomatic remission.
Complete symptomatic remission was defined as a SF subscore of 0 and RB subscore of 0. At Week 4, a greater proportion of patients treated with Velsipity compared to placebo achieved complete symptomatic remission (12% vs. 4%).
Stool frequency and rectal bleeding subscores.
Decreases in SF and RB subscores were observed as early as Week 2 in patients treated with Velsipity compared to placebo.
Cessation of rectal bleeding.
A greater proportion of patients achieved an RB subscore of 0 as early as Week 4 with Velsipity compared with placebo (47% vs. 25%).
Endoscopic and histologic assessment.
Normalisation of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. A greater proportion of patients treated with Velsipity compared to placebo achieved endoscopic remission by Week 12 (17% vs. 8%).
Endoscopic remission and Geboes histologic score < 2.0 (indicating no neutrophils in crypts or lamina propria and no increase in eosinophil, no crypt destruction, and no erosions, ulcerations, or granulation tissue) were achieved by a greater proportion of patients treated with Velsipity compared to placebo at Week 12 (10% vs. 5%).
When defined as ES ≤ 1 and Geboes ≤ 3.1 (indicating neutrophil infiltration in < 5% of crypts, no crypt destruction, and no erosions, ulcerations, or granulation tissue), a greater proportion of patients treated with Velsipity compared to placebo achieved histologic-endoscopic mucosal improvement at Week 12 (29% vs. 12%).
Abdominal pain and bowel urgency.
At Week 12, a greater proportion of patients treated with Velsipity compared to placebo had absence of abdominal pain (32% vs. 18%) and absence of bowel urgency (21% vs. 12%).
Inflammatory bowel disease questionnaire (IBDQ).
Patients treated with Velsipity compared to placebo demonstrated greater improvement from baseline in the total and all 4 domain scores of the IBDQ (bowel symptoms, systemic function, emotional function, and social function) at Week 12.
5.2 Pharmacokinetic Properties
Absorption.
Following etrasimod single oral dosing, Cmax and AUC increased approximately dose proportionally in the dose range studied (0.1 mg to 5 mg). Following multiple dosing, mean Cmax and AUC increased slightly more than dose proportional from 0.7 mg to 2 mg.
Steady state plasma concentrations are reached within 7 days following 2 mg once daily dosing, with a mean Cmax of 113 nanogram/mL and AUCtau of 2163 h*nanogram/mL. Steady state etrasimod accumulation is approximately 2- to 3-fold greater than single dose.
The pharmacokinetics of Velsipity is similar in healthy subjects and subjects with UC.
The time (Tmax) to reach maximum plasma concentrations (Cmax) after oral administration of immediate release oral dosage forms of etrasimod is approximately 4 hours (range 2 - 8 hours). Etrasimod absorption is extensive, based on high permeability and observation of relatively little intact etrasimod eliminated in the feces (11.2% of administered radioactive dose). Steady state exposure was reached within 7 days of dose initiation of etrasimod.
Effect of food.
Food intake can result in slightly delayed absorption (the median Tmax increased by 2 hours). Food does not have an effect on etrasimod exposure measures (Cmax and AUC); therefore, Velsipity can be administered without regard to meals.
Distribution.
Etrasimod distributes to body tissues with a mean oral volume (Vz/F) of distribution of 66 L. Etrasimod is highly protein bound, 97.9% to human plasma protein and mainly distributed in the plasma fraction of whole blood.
Metabolism.
Etrasimod is extensively metabolised by oxidation and dehydrogenation involving CYP2C8, CYP2C9, and CYP3A4, and with minor contribution via CYP2C19 and CYP2J2. UGTs and sulfotransferases are also involved in the metabolism of etrasimod. Unchanged etrasimod is the main circulating component in plasma.
Excretion.
After oral administration, the apparent steady state oral clearance (CL/F) was approximately 1 L/h. The mean plasma elimination half-life (t1/2) of etrasimod is approximately 30 hours.
Etrasimod is primarily eliminated hepatically with 82% recovery of total radioactive dose in the faeces and 4.89% in the urine. Unchanged etrasimod was only detected in faeces, but not in urine.
Specific populations.
Male and female patients.
Sex or weight have no clinically significant influence on etrasimod pharmacokinetics.
Racial or ethnic groups.
No clinically relevant pharmacokinetic differences were observed between Japanese, Chinese, and Caucasian subjects.
Paediatric population.
A population pharmacokinetics analysis predicted similar etrasimod exposures in adult and older adolescent (16 to < 18 years old) patients with UC.
The safety and efficacy of Velsipity in children and adolescents below the age of 16 years have not been established.
Elderly.
Population pharmacokinetic analyses showed that age did not have an effect on the pharmacokinetics of etrasimod in patients over 65 years of age. There is no meaningful difference in the pharmacokinetics in elderly patients compared to younger patients.
Renal impairment.
No dose adjustments are needed in patients with renal impairment as Cmax and AUC were comparable between subjects with severe renal impairment (comprised of subjects with eGFR ≤ 29 mL/min) and subjects with normal renal function. The effect of hemodialysis on the pharmacokinetics of etrasimod was not evaluated.
Hepatic impairment.
No dose adjustments are needed in patients with mild to moderate hepatic impairment. Etrasimod is not recommended in severe hepatic impairment. The total etrasimod AUC parameters are 13% and 29% higher in subjects with mild and moderate hepatic impairment, respectively, compared with subjects with normal liver function for the 2 mg single dose studied. The unbound etrasimod AUC in subjects with mild and moderate hepatic impairment was similar to subjects with normal liver function. The modest increased etrasimod AUC in subjects with moderate hepatic impairment is not expected to be clinically significant.
5.3 Preclinical Safety Data
Genotoxicity.
Etrasimod was negative in a battery of in vitro (Ames, chromosomal aberration in human peripheral blood lymphocytes) and in vivo (rat micronucleus) assays.
Carcinogenicity.
Oral carcinogenicity studies of etrasimod were conducted in mice and rats. In mice administered etrasimod for up to 104 weeks, there was an increase in hemangiosarcoma and hemangioma at 6 and 20 mg/kg/day in males and females. Systemic exposure at the no-observed-effect-level (NOEL) of 2 mg/kg/day was approximately 22 times that in humans at the MRHD of 2 mg/day. In rats, oral administration of etrasimod (up to 20 mg/kg/day) for up to 91 weeks, did not result in an increase in tumours. Plasma etrasimod exposure (AUC) at the highest dose tested is approximately 90 and 200 (males and females, respectively) times that in humans at the MRHD of 2 mg/day.6 Pharmaceutical Particulars
6.1 List of Excipients
Velsipity tablets contain the following inactive ingredients: magnesium stearate, mannitol, microcrystalline cellulose, sodium starch glycollate type A, Opadry II complete film coating system 85F110190-CN green (ID: 144806).
See Section 2 Qualitative and Quantitative Composition.
6.2 Incompatibilities
See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Velsipity 2 mg film coated tablets are supplied in:
High-density polyethylene (HDPE) bottles closed with a child-resistant polypropylene cap and packaged inside an outer carton each containing 30 tablets.
Aluminum strip with integrated desiccant layer and backing, containing 7, 28 or 98 tablets.
Not all presentations may be available.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy for safe disposal.
6.7 Physicochemical Properties
Chemical structure.
The chemical structure of etrasimod L-arginine:

CAS number.
1206123-37-6.7 Medicine Schedule (Poisons Standard)
S4 (Prescription Only Medicine).
Summary Table of Changes
