What is in this leaflet
Read all of this leaflet carefully before you start taking this medicine.
This leaflet answers some of the common questions about VEMLIDY tablets. It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist about your medical condition or treatment.
If you have questions, please ask your doctor or your pharmacist.
Keep this leaflet with your VEMLIDY medicine. You may need to read it again.
What VEMLIDY is used for
VEMLIDY is used for the treatment of chronic (long-lasting) hepatitis B virus (HBV) in adults.
VEMLIDY contains the active substance tenofovir alafenamide. This active substance is an antiviral medicine, known as a nucleotide reverse transcriptase inhibitor (NtRTI).
How VEMLIDY works in the treatment of Chronic Hepatitis B (CHB)
VEMLIDY works by interfering with the normal working of an enzyme (DNA polymerase) that is essential for the HBV to reproduce itself. VEMLIDY may help lower the amount of hepatitis B virus in your body by lowering the ability of the virus to multiply and infect new liver cells and can improve the inflammation and scar tissue caused by the hepatitis B virus in your liver. Lowering the amount of virus in your body may reduce the chance of developing cirrhosis, liver failure and liver cancer.
We do not know how long VEMLIDY may help treat your hepatitis. Sometimes viruses change in your body and medicines no longer work. This is called drug resistance.
It is not known if VEMLIDY is safe and effective for treatment of HBV in children or adolescents under 18 years of age.
Before you take VEMLIDY
When you must not take it
Do not take VEMLIDY if you have an allergy to:
- any medicine containing tenofovir alafenamide
- any of the ingredients in VEMLIDY listed in the product description section of this leaflet.
Some of the following symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- chest pain or tightness
- fainting
- rash, itching or hives on the skin
If you have any of the above symptoms soon after taking your medicine, DO NOT TAKE ANY MORE VEMLIDY and tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital. These are very serious effects. If you have them, you may have a serious allergic reaction. You may need urgent medical attention or hospitalisation.
Do not take VEMLIDY if you are taking any medicine that contains:
- tenofovir alafenamide
- tenofovir disoproxil fumarate
- adefovir dipivoxil
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have:
- Human Immunodeficiency Virus (HIV) infection. Your doctor may test you for HIV infection before starting VEMLIDY. If you have HIV and take VEMLIDY, the HIV virus may develop resistance and become harder to treat.
- any other medical conditions.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell him/her before you start taking VEMLIDY.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and VEMLIDY may interfere with each other. These include:
- rifampicin, rifapentine, rifabutin (antibiotics used to treat infections, including tuberculosis);
- St. John’s Wort (Hypericum perforatum – herbal medicine used to treat depression);
- carbamazepine, phenytoin, phenobarbital, oxcarbazepine (medicines used to treat epilepsy and prevent seizures).
- itraconazole, ketoconazole (antifungals used to treat infections)
These medicines may be affected by VEMLIDY or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
It is very important to let your doctor or pharmacist know what medications, herbal supplements, or vitamins you are taking.
Know the medicines you take. Keep a list of medicines and show it to your doctor and pharmacist when you get a new medicine. Your doctor and your pharmacist can tell you if you can take these medicines with VEMLIDY.
How to take VEMLIDY
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box or bottle, ask your doctor or pharmacist for help.
How much to take
Take VEMLIDY exactly as your doctor tells you to take it.
The usual dose is one VEMLIDY tablet orally, once daily.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it
Continue taking your medicine for as long as your doctor tells you. This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
When your VEMLIDY supply starts to run low, get more from your pharmacy. This is very important because your HBV infection may get worse (flare-up) if you stop taking VEMLIDY.
If you forget to take it
Do not miss a dose of VEMLIDY.
If you forget to take VEMLIDY, take your missed dose right away unless it is almost time for your next dose.
Do not take a double dose to make up for a forgotten dose. Continue with your regular dosing schedule.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre: 13 11 26 (Australia) and 0800 764 766 (New Zealand) or go to the Accident and Emergency department at your nearest hospital if you think you or anyone else may have taken too many VEMLIDY tablets. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using VEMLIDY
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking VEMLIDY.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor’s appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take VEMLIDY to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects.
Do not take VEMLIDY after the expiry or “use by” date (EXP) printed on the bottle. If you take it after the expiry date has passed, it may not work as well.
Do not take VEMLIDY if the packaging is torn or shows signs of tampering.
Things to be careful of
Be careful driving or operating machinery until you know how VEMLIDY affects you.
Side Effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking VEMLIDY.
This medicine helps most people with chronic HBV infection, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- nausea
This is not a complete list of side effects of VEMLIDY.
Check with your doctor as soon as possible if you have any problems while taking VEMLIDY, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Ask your doctor or pharmacist if you don’t understand anything in this list.
After taking VEMLIDY
Storage
Keep your VEMLIDY tablets in the bottle with the cap tightly closed until you take them. If you take the tablets out of the bottle they may not keep well.
Keep VEMLIDY tablets in a cool, dry place where it stays below 30 °C.
Do not store VEMLIDY or any other medicine in a bathroom or near a sink. Do not leave VEMLIDY in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product Description
What it looks like
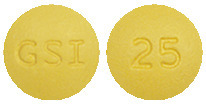
VEMLIDY tablets are round and yellow in colour. Each tablet has “GSI” on one side and “25” on the other side of the tablet.
VEMLIDY tablets are supplied in bottles containing 30 tablets.
Ingredients
VEMLIDY contains 25 mg of tenofovir alafenamide as the active ingredient.
It also contains:
- magnesium stearate
- croscarmellose sodium
- lactose monohydrate
- microcrystalline cellulose
- iron oxide yellow
- polyethylene glycol
- polyvinyl alcohol
- purified talc
- titanium dioxide
Supplier
VEMLIDY is supplied by:
Australia
Gilead Sciences Pty Ltd
Level 6, 417 St Kilda Road
Melbourne, Victoria 3004
New Zealand
c/- Grant Thornton New Zealand Limited,
L4, 152 Fanshawe Street
Auckland 1010
VEMLIDY and GSI are trademarks of Gilead Sciences, Inc. or one of its related companies. Other brands listed are trademarks of their respective owners and are not trademarks of Gilead Sciences, Inc.
This leaflet was prepared in February 2018.
AUST R 274395
Published by MIMS January 2019

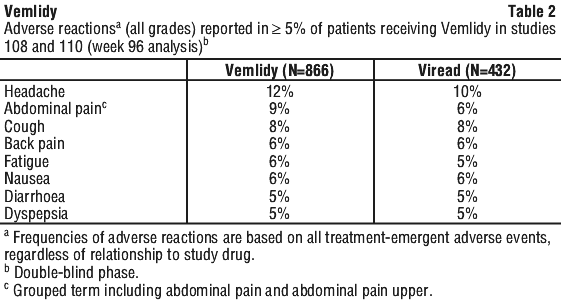 Additional adverse reactions occurring in greater than 1% to less than 5% of patients in Studies 108 and 110 included vomiting, rash, and flatulence.
Additional adverse reactions occurring in greater than 1% to less than 5% of patients in Studies 108 and 110 included vomiting, rash, and flatulence.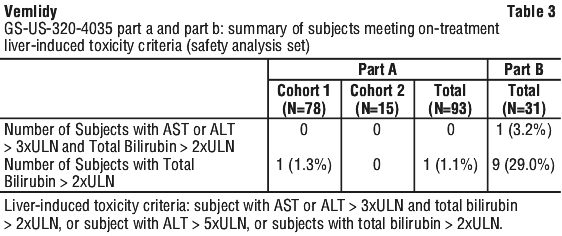
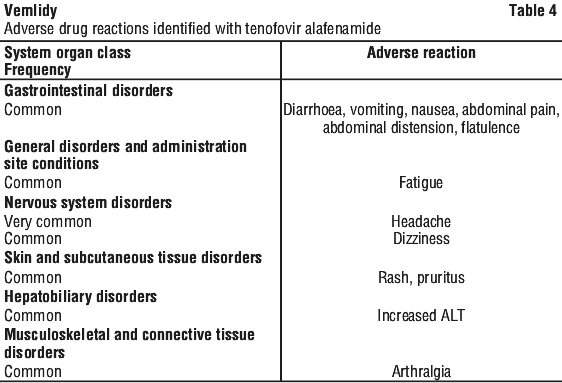
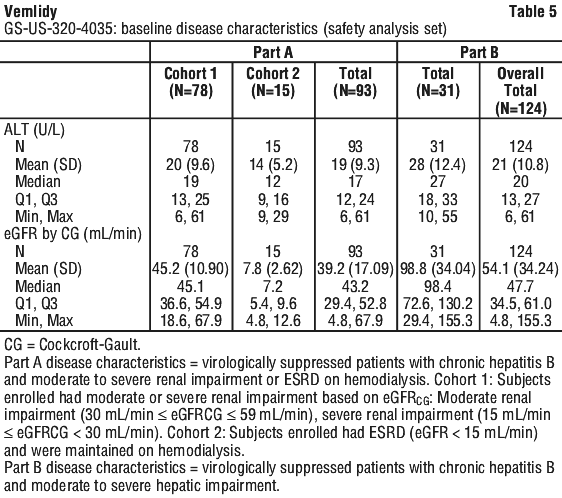
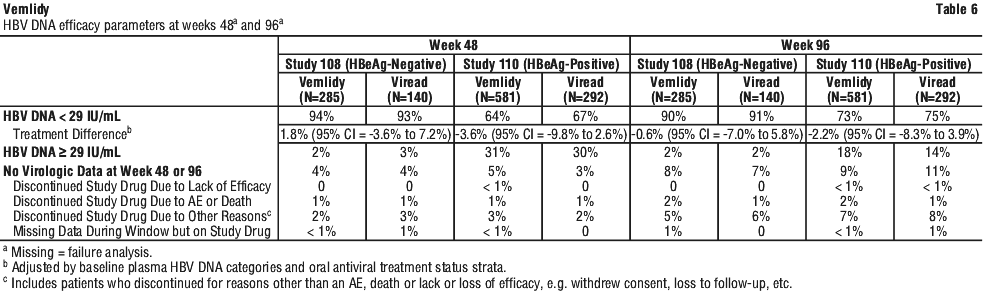 Vemlidy met the non-inferiority criteria in achieving HBV DNA less than 29 IU/mL when compared to Viread at Week 48. At Week 96, similar efficacy was demonstrated with Vemlidy compared to Viread.
Vemlidy met the non-inferiority criteria in achieving HBV DNA less than 29 IU/mL when compared to Viread at Week 48. At Week 96, similar efficacy was demonstrated with Vemlidy compared to Viread.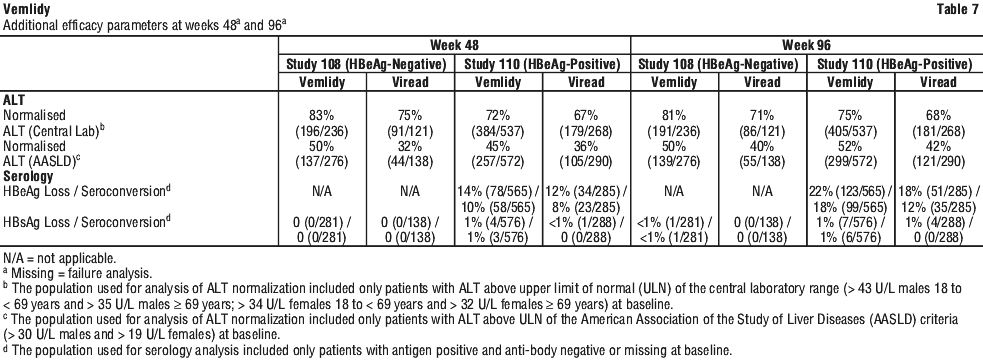
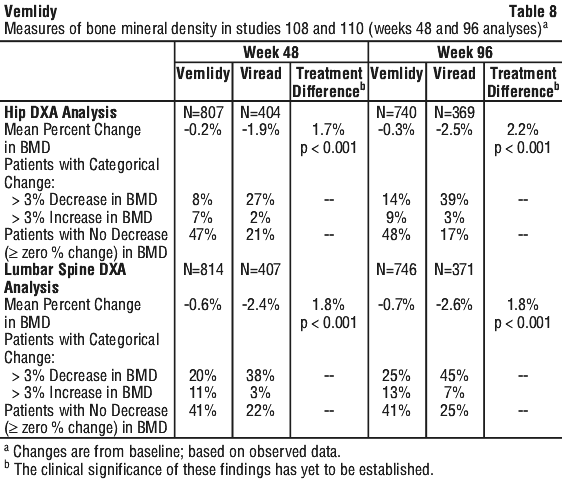 In patients who remained on blinded treatment beyond Week 96 in Studies 108 and 110, mean percentage change in BMD as assessed by DXA in each group at Week 120 was similar to that at Week 96. In the open-phase of Studies 108 and 110, mean percentage change in BMD from Week 96 to Week 120 in patients who remained on Vemlidy was +0.6% at the lumbar spine and 0% at the total hip, compared to +1.7% at the lumbar spine and +0.6% at the total hip in those who switched from Viread to Vemlidy at Week 96.
In patients who remained on blinded treatment beyond Week 96 in Studies 108 and 110, mean percentage change in BMD as assessed by DXA in each group at Week 120 was similar to that at Week 96. In the open-phase of Studies 108 and 110, mean percentage change in BMD from Week 96 to Week 120 in patients who remained on Vemlidy was +0.6% at the lumbar spine and 0% at the total hip, compared to +1.7% at the lumbar spine and +0.6% at the total hip in those who switched from Viread to Vemlidy at Week 96. In patients who remained on blinded treatment beyond Week 96 in Studies 108 and 110, change from baseline in renal laboratory parameter values in each group at Week 120 were similar to those at Week 96. In the open-label phase of Studies 108 and 110, the mean change (±SD) in serum creatinine from Week 96 to Week 120 was -0.002 (0.10) mg/dL in those who remained on Vemlidy, compared to -0.008 (0.09) mg/dL in those who switched from Viread to Vemlidy at Week 96. In the open-label phase, the median change in eGFR from Week 96 to Week 120 was -0.6 mL/min in patients who remained on Vemlidy, compared to +1.8 mL/min patients who switched from Viread to Vemlidy at Week 96.
In patients who remained on blinded treatment beyond Week 96 in Studies 108 and 110, change from baseline in renal laboratory parameter values in each group at Week 120 were similar to those at Week 96. In the open-label phase of Studies 108 and 110, the mean change (±SD) in serum creatinine from Week 96 to Week 120 was -0.002 (0.10) mg/dL in those who remained on Vemlidy, compared to -0.008 (0.09) mg/dL in those who switched from Viread to Vemlidy at Week 96. In the open-label phase, the median change in eGFR from Week 96 to Week 120 was -0.6 mL/min in patients who remained on Vemlidy, compared to +1.8 mL/min patients who switched from Viread to Vemlidy at Week 96.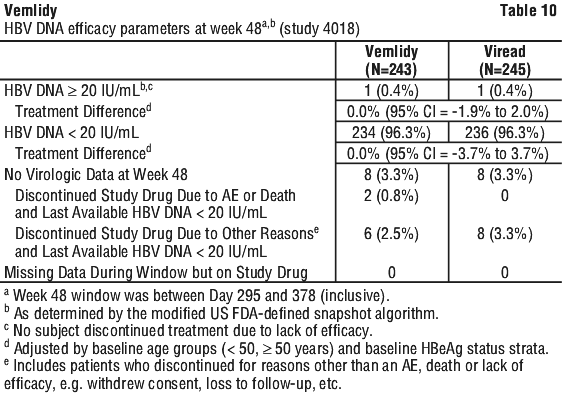 Vemlidy was noninferior in the proportion of subjects with HBV DNA ≥ 20 IU/mL at Week 48 when compared to Viread as assessed by the modified FDA Snapshot algorithm. Treatment outcomes (HBV DNA < 20 IU/mL by missing=failure) at Week 48 between treatment groups were similar across subgroups by age, sex, race, baseline HBeAg status, and ALT. See Table 11.
Vemlidy was noninferior in the proportion of subjects with HBV DNA ≥ 20 IU/mL at Week 48 when compared to Viread as assessed by the modified FDA Snapshot algorithm. Treatment outcomes (HBV DNA < 20 IU/mL by missing=failure) at Week 48 between treatment groups were similar across subgroups by age, sex, race, baseline HBeAg status, and ALT. See Table 11.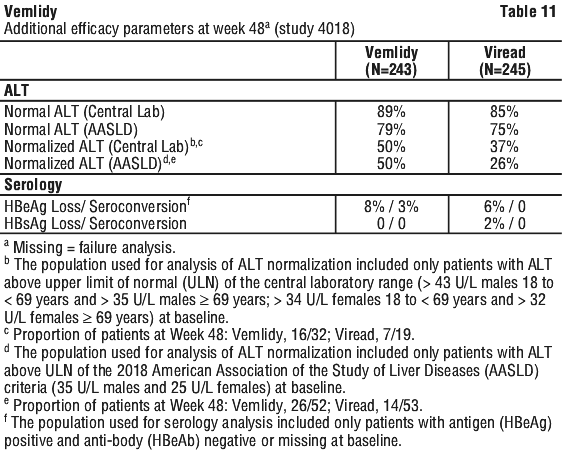
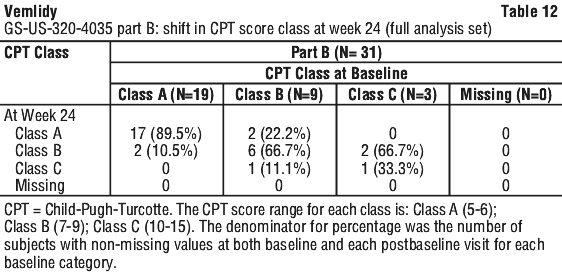

 Tenofovir alafenamide fumarate is a white to off-white or tan powder with a solubility of 4.7 mg per mL in water at 20°C.
Tenofovir alafenamide fumarate is a white to off-white or tan powder with a solubility of 4.7 mg per mL in water at 20°C.