

What is in this leaflet
This leaflet answers some common questions about Venclexta.
It does not contain all the available information. It does not take the place of talking to your doctor, nurse or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor, nurse or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
Warning
Tumour Lysis Syndrome (TLS), which can be fatal, has rarely occurred in patients receiving Venclexta.
Ensure you follow your doctor's instructions carefully especially when you start treatment with Venclexta. This includes keeping all of your appointments, including blood tests set up for you each week at the start of your treatment. The changes in your blood that could lead to TLS may show no symptoms.
Tell your doctor, nurse or pharmacist if you have or have had kidney problems, as this can increase the risk of TLS.
What Venclexta is used for
Venclexta is used, in combination with rituximab or alone, to treat a condition called "Chronic Lymphocytic Leukaemia (CLL)".
- CLL is a type of cancer affecting white blood cells called "B lymphocytes" and may also involve the lymph nodes, which are glands throughout the body that contain white blood cells. In CLL, the B cells multiply too quickly and live too long, so that there are too many of them in the blood.
Venclexta is used, in combination with azacitidine or with low dose cytarabine, to treat a condition called "Acute Myeloid Leukaemia (AML)". This medicine has provisional approval in Australia for AML. The decision to approve this medicine has been made on the basis of promising results from preliminary studies. More evidence is required to be submitted when available to fully confirm the benefit and safety of the medicine for this use.
- AML is a type of cancer affecting cells in the blood and bone marrow called "myeloid blasts". In AML, changes in these cells prevent them from turning into mature blood cells, resulting in too many of them and too few mature blood cells, platelets and other white blood cells in the blood.
The active substance in this medicine is called venetoclax.
Venclexta works by blocking a protein in the body ("BCL-2") that helps these cancer cells survive. Blocking this protein helps to kill and reduce the number of cancer cells, and may slow the spread of the disease.
Ask your doctor, nurse or pharmacist if you have any questions about why it has been prescribed for you. Your doctor may have prescribed it for another purpose.
This medicine is available only with a doctor’s prescription.
Before you take Venclexta
When you must not take it
Do not take Venclexta if you have an allergy to:
- any medicine containing venetoclax
- any of the ingredients listed at the end of this leaflet.
Some symptoms of an allergic reaction include
- hives, skin rash or itching
- shortness of breath, wheezing, difficulty breathing or a tight feeling in your chest
- swelling of the face, lips or tongue, which may cause difficulty in swallowing or breathing.
For patients with CLL, do not take Venclexta if you are taking any medicine that contains:
- ketoconazole, posaconazole, voriconazole or itraconazole, medicines used for fungal infections
- clarithromycin, a medicine used to treat bacterial infections
- conivaptan, a medicine used to treat low sodium levels
- ritonavir, indinavir, lopinavir or telaprevir, medicines used to treat HIV or HCV (hepatitis C virus) infections.
Do not take it after the expiry date printed on the pack or if the packaging is damaged or shows signs of tampering. If it has expired or is damaged return it to your pharmacist for disposal.
If you use this medicine after the expiry date has passed it may not work as well.
Before you start to take it
Tell your doctor or pharmacist if you have or have had any of the following medical conditions:
- heart, kidney or liver problems
- an infection.
If you have not told your doctor or pharmacist about any of the above, tell them before you take Venclexta.
Tell your doctor if you recently received or are scheduled to receive a vaccine. Certain vaccines may cause infections and should not be given while taking Venclexta.
Do not take Venclexta if you are pregnant or plan to become pregnant. Venclexta should not be used during pregnancy. Your doctor may conduct a pregnancy test and discuss contraception options with you before you start taking your medicine.
Do not take Venclexta if you are breastfeeding or plan to breastfeed. It is not known whether this medicine passes into the breast milk.
Your doctor will discuss the risks and benefits of using this medicine if you are breastfeeding.
Tell your doctor or pharmacist if you have any allergies to any other medicines, foods, preservatives or dyes.
If you have not told your doctor or pharmacist about any of the above, tell them before you take Venclexta.
Use in Children
Do not give Venclexta to children and adolescents under 18 years of age. The safety and effectiveness of Venclexta in children and adolescents have not yet been established.
Tumour Lysis Syndrome
Venclexta can cause Tumour Lysis Syndrome, a very serious side effect that can be fatal.
Some people having treatment for cancer can develop Tumour Lysis Syndrome (TLS), which is caused by the fast breakdown of cancer cells. As the cancer cells are destroyed, they break open and what is inside the cancer cell (uric acid, potassium, phosphorus) gets into the blood. This can lead to changes in kidney function, sudden kidney failure or even death.
To help prevent TLS, drink 6-8 glasses (approximately 1.5 - 2 litres total) of water each day, especially starting 2 days before and on the day of your first dose of Venclexta, and every time the dose is increased. It is important that you continue to remain hydrated throughout your treatment.
Before you start your treatment, your doctor will do blood tests and a scan (for example, a CT scan) to see if you are at risk of developing TLS. It is important for you to keep your scheduled appointments for blood tests. The changes in your blood that could lead to TLS may have no symptoms.
If the following symptoms occur, contact your doctor immediately:
- fever or chills
- feeling or being sick (nausea or vomiting)
- feeling confused
- feeling short of breath
- irregular heart beat
- dark or cloudy urine
- fits or seizures
- feeling unusually tired
- muscle pain or joint discomfort.
TLS is most likely to occur when you are first starting your treatment.
Follow all instructions given to you by your doctor.
Your doctor may give you medicines (such as allopurinol) to help prevent the build-up of uric acid in your body before you start treatment with Venclexta.
Taking other medicines
Do not take Venclexta and talk to your doctor if you take a medicine that contains:
- ketoconazole, posaconazole, voriconazole or itraconazole, medicines used for fungal infections
- clarithromycin, a medicine used to treat bacterial infections
- conivaptan, a medicine used to treat low sodium levels
- ritonavir, indinavir, lopinavir or telaprevir, medicines used to treat HIV or HCV infections.
For patients with AML, your doctor may choose to reduce the starting dose of Venclexta or provide other advice on how your treatment should be managed.
Serious or life-threatening effects can occur when Venclexta is taken with certain medicines, when treatment starts and during the time when the dose is increased.
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food store.
Your doctor may need to stop certain medicines when you first start taking Venclexta and when your dose is gradually increased to the full standard dose.
If your are taking certain medications that can interact with Venclexta, your doctor may also choose to reduce the starting dose.
Some medicines and Venclexta may interfere with each other.
Tell your doctor if you are taking:
- fluconazole, a medicine used to treat fungal infections
- ciprofloxacin, erythromycin, azithromycin, nafcillin or rifampicin, medicines called antibiotics that are used to treat bacterial infections
- carbamazepine or phenytoin, medicines used to prevent seizures or to treat epilepsy
- efavirenz or etravirine, medicines used to treat HIV infection
- captopril, carvedilol, felodipine, ranolazine, bosentan, verapamil or diltiazem, medicines used to treat blood pressure or angina
- modafinil, a medicine used to treat a sleep disorder known as narcolepsy
- herbal medicines: St. John's Wort (Hypericum perforatum) or quercetin
- warfarin, a medicine used to thin the blood
- amiodarone, quinidine, ticagrelor, digoxin or dronedarone, medicines used to treat heart failure or heart rhythm problems
- everolimus or sirolimus, medicines used to treat cancer and patients who have had organ transplants
- ciclosporin, a medicine used to suppress the immune system.
These medicines may be affected by Venclexta, or may affect how well it works. You may need to use different amounts of your medicine, or take different medicines.
Your doctor or pharmacist has more information on medicines to be careful with or to avoid while taking Venclexta.
How to take Venclexta
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
It is important you follow the instructions your doctor provides about how to take Venclexta and for how long. Not taking the medicine as directed or stopping the medicine too early may cause you to not respond to the medicine, may affect your response to future treatments, and may increase the risk of side effects
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
Take Venclexta exactly as your doctor has prescribed. Your doctor will tell you how many tablets to take and at what time of the day.
CLL
For patients with CLL, you will begin treatment with Venclexta at a low dose for 1 week. Your doctor will gradually increase the dose over the next four weeks to the full standard dose.
- Week 1: The starting dose is 20 mg (two 10 mg tablets) once a day
- Week 2: The dose is 50 mg (one 50 mg tablet) once a day
- Week 3: The dose is 100 mg (one 100 mg tablet) once a day
- Week 4: The dose 200 mg (two 100 mg tablets) once a day
- Week 5 and onwards: The dose is 400 mg (four 100 mg tablets) once a day. You will stay on the 400 mg daily dose, which is the standard dose, for as long as your doctor determines is necessary.
AML
For patients with AML taking Venclexta in combination with azacitidine:
You will begin treatment with Venclexta at a low dose. Your doctor will gradually increase the dose over the next three days to the full standard dose.
- The starting dose is 100 mg (one 100 mg tablet) once a day for 1 day
- Day 2: The dose will be increased to 200 mg (two 100 mg tablets) once a day for 1 day
- Day 3 and onwards: The dose will be increased to 400 mg (four 100 mg tablets) once a day. You will stay on the 400 mg daily dose, which is the standard dose, for as long as your doctor determines it necessary.
For patients with AML taking Venclexta in combination with lowdose cytarabine:
You will begin treatment with Venclexta at a low dose. Your doctor will gradually increase the dose over the next four days to the full standard dose.
- The starting dose is 100 mg (one 100 mg tablet) once a day for 1 day
- Day 2: The dose will be increased to 200 mg (two 100 mg tablets) once a day for 1 day
- Day 3: The dose will be increased to 400 mg (four 100 mg tablets) once a day for 1 day
- Day 4 and onwards: The dose will be increased to 600 mg (six 100 mg tablets) once a day. You will stay on the 600 mg daily dose, which is the standard dose, for as long as your doctor determines it necessary.
Ask your doctor or pharmacist if you are unsure.
Your doctor may change the dose of Venclexta you take based on test results during treatment.
Follow all of the instructions of your doctor and pharmacist. If you are being treated for CLL, also refer to the Quick Start Guide provided with your monthly starting pack for instructions on which tablets to take and when to take them.
If you are not sure how much and when to take it, ask your doctor or pharmacist.
How to take it
Swallow the tablets whole with a glass of water.
Do not chew, crush, or break the tablets.
Do not drink grapefruit juice, eat grapefruit, starfruit or Seville oranges or marmalades while you are taking Venclexta. These products may increase the amount of Venclexta in your blood.
When to take it
Take Venclexta during or immediately after a meal, at about the same time every day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps control your condition, but it does not cure it. It is important to keep taking your medicine even if you feel well.
If you become pregnant while taking Venclexta, you should immediately stop taking it and tell your doctor.
If you vomit after taking it
Do not take any additional dose that day. Take the dose at the usual time the next day.
If you forget to take it
It is important not to miss a dose of this medicine.
If you do miss a dose of Venclexta:
- By less than 8 hours
- Take the missed dose with food as soon as possible. Take your next dose the following day as usual. - By more than 8 hours
- Do not take the dose that day. Take your next dose at your usual time the next day.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your doctor or pharmacist for advice.
If you take too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre (telephone 13 11 26), or go to Accident and Emergency at your nearest hospital, if you think you or anyone else may have taken too much Venclexta. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking Venclexta
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Venclexta.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon that you are taking this medicine.
Tell your doctor immediately if you become pregnant while taking Venclexta.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Tell your doctor if, for any reason, you have not used Venclexta as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily. You may also be at a greater risk of experiencing side effects if you do not take Venclexta as directed.
Things you must not do
Do not use this medicine to treat any other complaints unless your doctor or pharmacist tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking Venclexta, or change the dosage, without checking with your doctor or pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how Venclexta affects you. The effect of Venclexta on your ability to drive or use machines is not known.
Be careful when drinking alcohol while you are taking this medicine. If you drink alcohol, symptoms such as nausea or vomiting may be worse.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Venclexta.
All medicines have some side effects. Sometimes they are serious, but most of the time they are not. You may need medical attention if you get some of the side-effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- feeling very tired
- feeling sick
- unusual weakness or lack of energy
- tiredness, headaches, being short of breath when exercising, dizziness and looking pale. These are symptoms of anaemia
- bruising or bleeding more than normal (e.g. blood in your urine or faeces, nose bleeds)
- diarrhoea and vomiting
- constipation
- decreased appetite
- dizziness
- headache
- cough
- shortness of breath, difficulty breathing or chest tightness.
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor or pharmacist as soon as possible if you notice any of the following:
- fever
- chills
- feeling weak or confused
- cough
- pain or burning feeling when passing urine.
These are possible signs of infection. Your doctor may check your blood count during treatment with Venclexta. Low white blood cell count (known as neutropenia) can increase your risk of infection.
Some infections can be very serious.
Tell your doctor immediately if you have signs of an infection while taking Venclexta.
Tell your doctor or pharmacist immediately if you notice any of the following:
- fever or chills
- feeling or being sick (nausea or vomiting)
- feeling confused
- feeling short of breath
- irregular heart beat
- dark or cloudy urine
- fits or seizures
- feeling unusually tired
- muscle pain or joint discomfort.
These symptoms can be associated with TLS. Refer to the section of this leaflet titled "Tumour Lysis Syndrome" for further information on TLS.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell.
After using Venclexta
Storage
Keep your tablets in the pack/bottle until it is time to take them.
Keep the medicine in a cool, dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom, near a sink, or on a windowsill. Do not leave it in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine, or the medicine has passed its expiry date, return any of the unused medicine to your pharmacist for disposal.
Product description
What it looks like
Venclexta tablets come in three strengths:
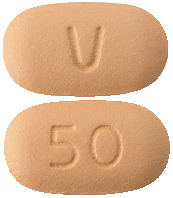
- Venclexta 10 mg film-coated tablets are pale yellow, round 6 mm diameter, with "V" on one side and "10" on the other,
- Venclexta 50 mg film-coated tablets are beige, oblong 14 mm long, with "V" on one side and "50" on the other,
- Venclexta 100 mg film-coated tablets are pale yellow, oblong 17 mm long, with "V" on one side and "100" on the other.
For patients with CLL, Venclexta is available as a Starting Pack, which is designed to provide you with the first four weeks of your dosing regimen.
Each starting pack is presented as a carton containing four weekly wallets:
- Week 1 (14 x 10 mg tablets)
- Week 2 (7 x 50 mg tablets)
- Week 3 (7 x 100 mg tablets)
- Week 4 (14 x 100 mg tablets)
The 100 mg strength is also available as a bottle (containing 120 or 180 tablets) and a blister pack (containing 7, 14 or 112 tablets).
Not all presentations may be available.
Ingredients
Active Ingredient
The active ingredient is venetoclax. Each film-coated tablet contains 10 mg, 50 mg or 100 mg venetoclax.
Inactive ingredients
- copovidone
- colloidal anhydrous silica
- polysorbate 80
- sodium stearylfumarate
- calcium hydrogen phosphate
- iron oxide yellow
- iron oxide red (50 mg tablet only)
- iron oxide black (50 mg tablet only)
- polyvinyl alcohol
- macrogol 3350
- purified talc
- titanium dioxide
Venclexta does not contain gluten, sucrose, tartrazine or any other azo dyes.
Supplier
Venclexta is supplied in Australia by:
AbbVie Pty Ltd
241 O'Riordan Street
Mascot NSW 2020
Australia
This leaflet was prepared in February 2020.
Australian Registration Number(s)
AUST R 267441, 267442, 267443, 267444, 267445.
Version 6
Published by MIMS April 2020


 Initiate azacitidine or low-dose cytarabine on cycle 1 day 1.
Initiate azacitidine or low-dose cytarabine on cycle 1 day 1.




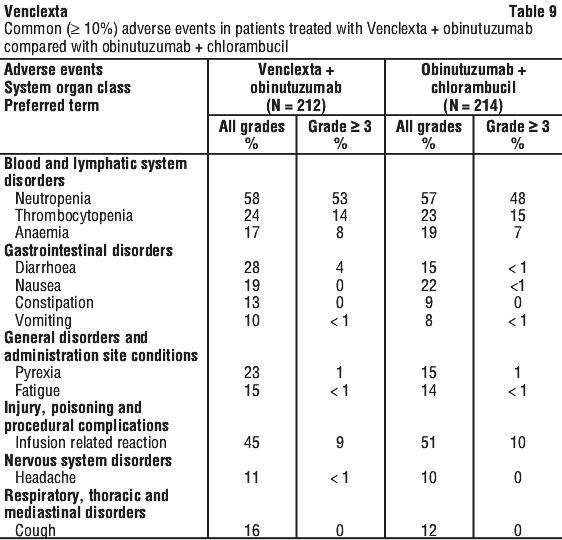 Other adverse reactions reported in the venetoclax + obinutuzumab arm are presented below:
Other adverse reactions reported in the venetoclax + obinutuzumab arm are presented below:
 Other adverse reactions reported in the Venclexta + ibrutinib arm are presented below:
Other adverse reactions reported in the Venclexta + ibrutinib arm are presented below: Other adverse reactions reported in the CAPTIVATE study are presented below:
Other adverse reactions reported in the CAPTIVATE study are presented below: Based on the existing safety profile of Venclexta, adverse reactions reported in the venetoclax + rituximab arm of MURANO that fall below the cut-off in Table 13 are presented below by MedDRA body system organ class and by frequency.
Based on the existing safety profile of Venclexta, adverse reactions reported in the venetoclax + rituximab arm of MURANO that fall below the cut-off in Table 13 are presented below by MedDRA body system organ class and by frequency. The most frequently reported serious adverse reactions (≥ 2%) unrelated to disease progression were pneumonia and febrile neutropenia.
The most frequently reported serious adverse reactions (≥ 2%) unrelated to disease progression were pneumonia and febrile neutropenia. Other adverse reactions (all grades) reported in the venetoclax + azacitidine arm are presented below:
Other adverse reactions (all grades) reported in the venetoclax + azacitidine arm are presented below: Other adverse drug reactions reported in the venetoclax + low-dose cytarabine arm are presented below:
Other adverse drug reactions reported in the venetoclax + low-dose cytarabine arm are presented below: At baseline, the median lymphocyte count was 55 x 109 cells/L in both study arms. On cycle 1 day 15, the median count decreased to 1.03 x 109 cells/L (range 0.2-43.4 x 109 cells/L) in the obinutuzumab + chlorambucil arm compared with 1.27 x 109 cells/L (range 0.2-83.7 x 109 cells/L) in the venetoclax + obinutuzumab arm.
At baseline, the median lymphocyte count was 55 x 109 cells/L in both study arms. On cycle 1 day 15, the median count decreased to 1.03 x 109 cells/L (range 0.2-43.4 x 109 cells/L) in the obinutuzumab + chlorambucil arm compared with 1.27 x 109 cells/L (range 0.2-83.7 x 109 cells/L) in the venetoclax + obinutuzumab arm. At the time of analysis, median overall survival (OS) had not been reached, with fewer than 10% of patients experiencing an event. The median duration of follow-up for OS was 28 months.
At the time of analysis, median overall survival (OS) had not been reached, with fewer than 10% of patients experiencing an event. The median duration of follow-up for OS was 28 months. Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 58% (126/216) in patients treated with venetoclax + obinutuzumab and 9% (20/216) in patients treated with obinutuzumab + chlorambucil.
Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 58% (126/216) in patients treated with venetoclax + obinutuzumab and 9% (20/216) in patients treated with obinutuzumab + chlorambucil. The Kaplan-Meier curve for investigator-assessed PFS is shown in Figure 1.
The Kaplan-Meier curve for investigator-assessed PFS is shown in Figure 1. The PFS benefit with venetoclax + obinutuzumab versus obinutuzumab + chlorambucil treatment was observed across all subgroups of patients evaluated (Figure 2).
The PFS benefit with venetoclax + obinutuzumab versus obinutuzumab + chlorambucil treatment was observed across all subgroups of patients evaluated (Figure 2).


 Across the high-risk CLL/SLL population (n = 123), including TP53 mutation (n = 9), 11q deletion (n = 38), or unmutated IgHV (n = 109), the treatment effect of venetoclax + ibrutinib was consistent, with a PFS HR of 0.23 [95% CI (0.13, 0.41)].
Across the high-risk CLL/SLL population (n = 123), including TP53 mutation (n = 9), 11q deletion (n = 38), or unmutated IgHV (n = 109), the treatment effect of venetoclax + ibrutinib was consistent, with a PFS HR of 0.23 [95% CI (0.13, 0.41)]. Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 49.1% (52/106) by next-generation sequencing (NGS) assay and 54.7% (58/106) by flow cytometry in patients treated with venetoclax + ibrutinib and, at the corresponding time point, was 12.4% (13/105) by NGS assay and 16.2% (17/105) by flow cytometry in patients treated with chlorambucil + obinutuzumab.
Twelve months after the completion of treatment, MRD negativity rates in peripheral blood were 49.1% (52/106) by next-generation sequencing (NGS) assay and 54.7% (58/106) by flow cytometry in patients treated with venetoclax + ibrutinib and, at the corresponding time point, was 12.4% (13/105) by NGS assay and 16.2% (17/105) by flow cytometry in patients treated with chlorambucil + obinutuzumab.
 At 3 months post-treatment in the fixed-duration cohort, 84 patients who were MRD negative in peripheral blood had matched bone marrow specimens; of these, 76 patients (90%) were MRD negative in both peripheral blood and bone marrow.
At 3 months post-treatment in the fixed-duration cohort, 84 patients who were MRD negative in peripheral blood had matched bone marrow specimens; of these, 76 patients (90%) were MRD negative in both peripheral blood and bone marrow. The median survival follow-up at the time of analysis was 23.8 months (range: 0.0 to 37.4 months).
The median survival follow-up at the time of analysis was 23.8 months (range: 0.0 to 37.4 months). At the time of primary analysis (data cutoff date 8 May 2017), 65 patients completed the 24 month venetoclax + rituximab treatment regimen without progression and 78 patients were still receiving venetoclax (+18 months of treatment). Of the 65 patients who remained progression free at 24 months, only 2 patients progressed after treatment completion. Twelve patients had a 3-month follow-up visit and remained progression free. Of the 12 patients, 5 were also assessed at 6-month follow-up and remained progression free.
At the time of primary analysis (data cutoff date 8 May 2017), 65 patients completed the 24 month venetoclax + rituximab treatment regimen without progression and 78 patients were still receiving venetoclax (+18 months of treatment). Of the 65 patients who remained progression free at 24 months, only 2 patients progressed after treatment completion. Twelve patients had a 3-month follow-up visit and remained progression free. Of the 12 patients, 5 were also assessed at 6-month follow-up and remained progression free. In total, 130 patients in the venetoclax + rituximab arm completed 2 years of Venclexta treatment without progression. For these patients, the 3-year PFS estimate post-treatment was 51% [95% CI: 40.2, 61.9].
In total, 130 patients in the venetoclax + rituximab arm completed 2 years of Venclexta treatment without progression. For these patients, the 3-year PFS estimate post-treatment was 51% [95% CI: 40.2, 61.9]. The Kaplan-Meier curve for overall survival is shown in Figure 5.
The Kaplan-Meier curve for overall survival is shown in Figure 5. The observed PFS benefit of venetoclax + rituximab compared with bendamustine + rituximab was consistently observed across all subgroups of patients evaluated, including high-risk patients with deletion 17p/TP53 mutation and/or unmutated IgVH (Figure 6).
The observed PFS benefit of venetoclax + rituximab compared with bendamustine + rituximab was consistently observed across all subgroups of patients evaluated, including high-risk patients with deletion 17p/TP53 mutation and/or unmutated IgVH (Figure 6).
 Among the patients, 37% (34/91) were fludarabine refractory, 81% (30/37) had unmutated IgVH, and 24% (19/80) had 11q deletion.
Among the patients, 37% (34/91) were fludarabine refractory, 81% (30/37) had unmutated IgVH, and 24% (19/80) had 11q deletion. Based on a later data cutoff (15 June 2017), which included an additional 51 patients enrolled in a safety expansion cohort, and investigator-assessed efficacy (N = 158), the median duration of response (DOR) was 36.2 months (95% CI: 27.2, NA). The median duration of progression-free survival (mPFS) was 28.2 months (95% CI: 23.4, 37.0).
Based on a later data cutoff (15 June 2017), which included an additional 51 patients enrolled in a safety expansion cohort, and investigator-assessed efficacy (N = 158), the median duration of response (DOR) was 36.2 months (95% CI: 27.2, NA). The median duration of progression-free survival (mPFS) was 28.2 months (95% CI: 23.4, 37.0). Among the patients, 70% were fludarabine refractory, 67% (22/33) had unmutated IgVH, 31% (18/58) had 11q deletion, and 24% (14/58) had 17p deletion.
Among the patients, 70% were fludarabine refractory, 67% (22/33) had unmutated IgVH, 31% (18/58) had 11q deletion, and 24% (14/58) had 17p deletion. For the 8 patients with SLL, the investigator-assessed ORR was 100%.
For the 8 patients with SLL, the investigator-assessed ORR was 100%. Efficacy data are presented with data cutoff date of 26 July 2017. Investigator-assessment of disease responses to venetoclax treatment are available for all 127 subjects (64 in the main cohort and 63 in the expansion cohort). The IRC assessments of disease responses are available for 123 of the 127 subjects.
Efficacy data are presented with data cutoff date of 26 July 2017. Investigator-assessment of disease responses to venetoclax treatment are available for all 127 subjects (64 in the main cohort and 63 in the expansion cohort). The IRC assessments of disease responses are available for 123 of the 127 subjects. The median duration of treatment with venetoclax for 127 patients was 14.3 months (range: 0.1 to 31.4 months).
The median duration of treatment with venetoclax for 127 patients was 14.3 months (range: 0.1 to 31.4 months). The dual primary endpoints of the study were overall survival (OS) measured from the date of randomisation to death from any cause and composite complete remission rate (complete remission + complete remission with incomplete blood count recovery; CR+CRi). The overall median follow-up at the time of analysis was approximately 20.5 months (range: < 0.1 to 30.7 months).
The dual primary endpoints of the study were overall survival (OS) measured from the date of randomisation to death from any cause and composite complete remission rate (complete remission + complete remission with incomplete blood count recovery; CR+CRi). The overall median follow-up at the time of analysis was approximately 20.5 months (range: < 0.1 to 30.7 months).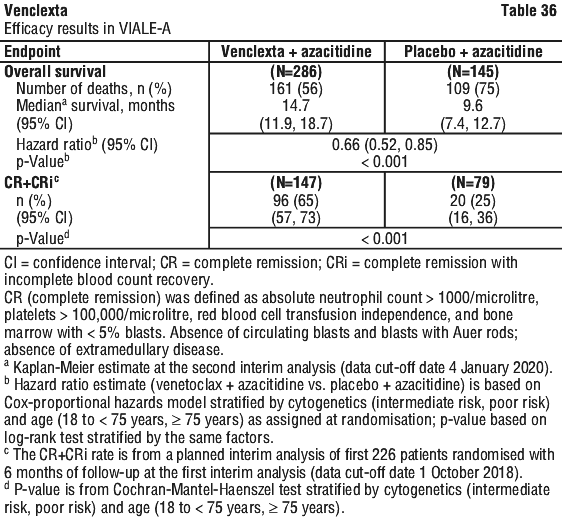
 Key secondary efficacy endpoints are presented in Table 37.
Key secondary efficacy endpoints are presented in Table 37.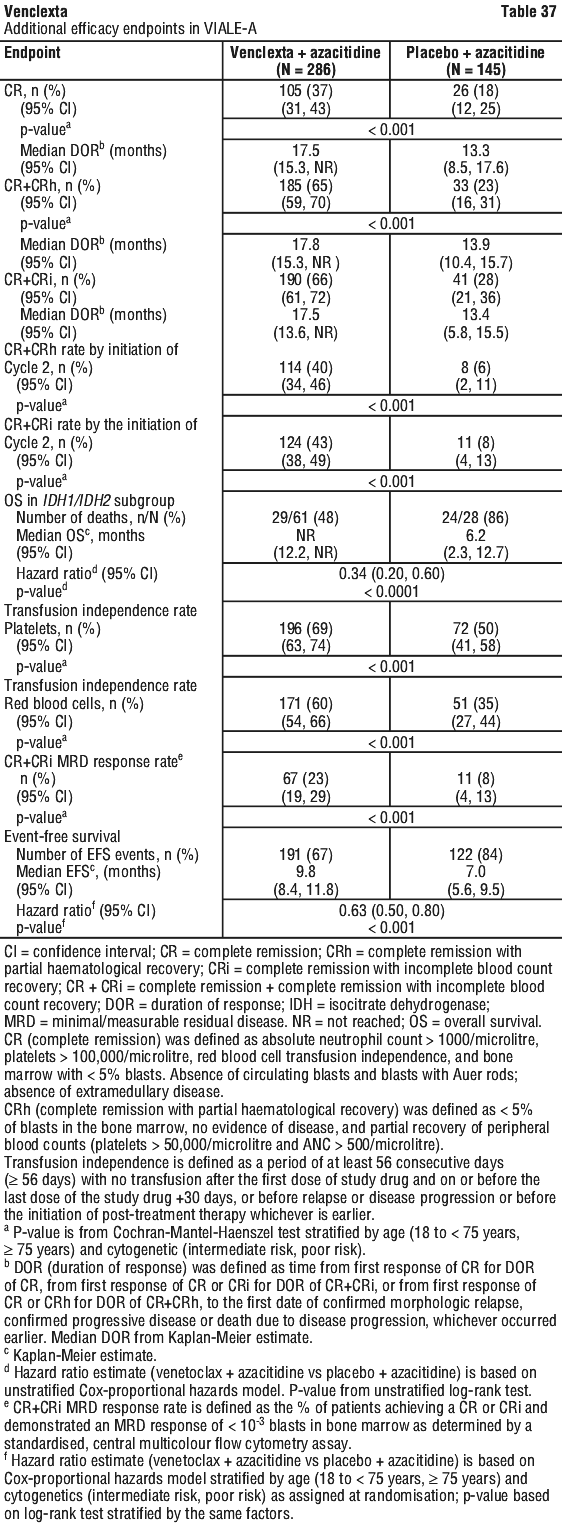 Of patients with FLT3 mutations, the CR+CRh rates were 66% (19/29; [95% CI: 46, 82]) and 18% (4/22; [95% CI: 5, 40]) in the venetoclax + azacitidine and placebo + azacitidine arms, respectively (Fisher's exact test p = 0.001). Of patients with FLT3 mutations, the CR+CRi rates were 72% (21/29; [95% CI: 53, 87]) and 36% (8/22; [95% CI: 17, 59]) in the venetoclax + azacitidine and placebo + azacitidine arms, respectively (Fisher's exact test p = 0.021).
Of patients with FLT3 mutations, the CR+CRh rates were 66% (19/29; [95% CI: 46, 82]) and 18% (4/22; [95% CI: 5, 40]) in the venetoclax + azacitidine and placebo + azacitidine arms, respectively (Fisher's exact test p = 0.001). Of patients with FLT3 mutations, the CR+CRi rates were 72% (21/29; [95% CI: 53, 87]) and 36% (8/22; [95% CI: 17, 59]) in the venetoclax + azacitidine and placebo + azacitidine arms, respectively (Fisher's exact test p = 0.021). At the time of the primary analysis for OS, patients had a median follow-up of 12 months (range: 0.1 to 17.6 months). The median OS in the venetoclax + low-dose cytarabine arm was 7.2 months (95% CI: 5.6, 10.1) and in the placebo with low-dose cytarabine arm was 4.1 months (95% CI: 3.1, 8.8). The hazard ratio was 0.75 (95% CI: 0.52, 1.07; p = 0.114). The Kaplan-Meier curve for OS is shown in Figure 8.
At the time of the primary analysis for OS, patients had a median follow-up of 12 months (range: 0.1 to 17.6 months). The median OS in the venetoclax + low-dose cytarabine arm was 7.2 months (95% CI: 5.6, 10.1) and in the placebo with low-dose cytarabine arm was 4.1 months (95% CI: 3.1, 8.8). The hazard ratio was 0.75 (95% CI: 0.52, 1.07; p = 0.114). The Kaplan-Meier curve for OS is shown in Figure 8. In an additional analysis for OS at which time patients had a median follow-up of 17.5 months (range: 0.1 to 23.5 months). The median OS in the venetoclax + low-dose cytarabine arm was 8.4 months (95% CI: 5.9, 10.1) and in the placebo + low-dose cytarabine arm was 4.1 months (95% CI: 3.1, 8.1). The hazard ratio was 0.70 (95% CI: 0.50, 0.99). The Kaplan-Meier curve for OS with 6 additional months of follow up is shown in Figure 9.
In an additional analysis for OS at which time patients had a median follow-up of 17.5 months (range: 0.1 to 23.5 months). The median OS in the venetoclax + low-dose cytarabine arm was 8.4 months (95% CI: 5.9, 10.1) and in the placebo + low-dose cytarabine arm was 4.1 months (95% CI: 3.1, 8.1). The hazard ratio was 0.70 (95% CI: 0.50, 0.99). The Kaplan-Meier curve for OS with 6 additional months of follow up is shown in Figure 9.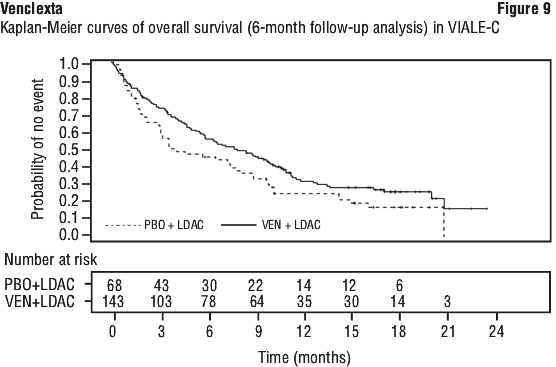 Efficacy results for secondary endpoints from the primary analysis are shown in Table 39.
Efficacy results for secondary endpoints from the primary analysis are shown in Table 39.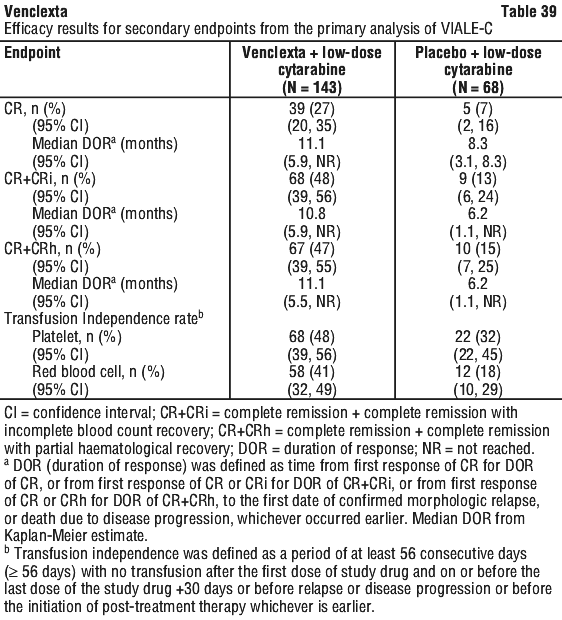 The CR+CRi rate by initiation of cycle 2 for venetoclax + low-dose cytarabine was 34% (95% CI: 27, 43) and for placebo + low-dose cytarabine was 3% (95% CI: 0.4, 10). The median time to first response of CR+CRi was 1.1 months (range: 0.8 to 4.7 months) with venetoclax + low-dose cytarabine treatment. The median time to best response of CR+CRi was 1.2 months (range: 0.8 to 5.9 months).
The CR+CRi rate by initiation of cycle 2 for venetoclax + low-dose cytarabine was 34% (95% CI: 27, 43) and for placebo + low-dose cytarabine was 3% (95% CI: 0.4, 10). The median time to first response of CR+CRi was 1.1 months (range: 0.8 to 4.7 months) with venetoclax + low-dose cytarabine treatment. The median time to best response of CR+CRi was 1.2 months (range: 0.8 to 5.9 months). Not all presentations may be marketed.
Not all presentations may be marketed. Empirical formula: C45H50ClN7O7S.
Empirical formula: C45H50ClN7O7S.