
What is in this leaflet
This leaflet answers some common questions about VFEND. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking VFEND against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What VFEND is used for
VFEND is used to treat fungal and yeast infections such as:
- invasive aspergillosis (as-pur-ji-losis), a fungal infection caused by a fungus called Aspergillus (as-pur-jilus), which usually begins in the respiratory tract (in the nose, sinuses or lungs). Aspergillus is harmless in most healthy people; however, in people with poor immune systems (such as people who have had organ transplants and people with cancer or HIV/AIDS) invasive aspergillosis can be serious and spread to other tissues and organs.
- serious Candida (can-did-da) infections, including Candida infections of the oesophagus (food pipe or gullet) and those that have spread into the blood stream or to other parts of the body.
- serious fungal infections caused by Scedosporium (ski-doe-spore-rium) species and Fusarium (few-saa-rium) species.
- other serious fungal infections in patients who do not respond to, or cannot tolerate, other antifungal medicines.
VFEND is also used to prevent invasive fungal infections in patients who are at risk of developing such infections.
This medicine belongs to a group of medicines called triazole antifungals.
This medicine works by preventing the growth of fungal and yeast organisms causing your infection.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
Before you start to use VFEND
When you must not use it
Do not use VFEND if you have ever had an allergy to:
- any medicine containing voriconazole.
- any of the ingredients listed at the end of this leaflet.
- any other similar medicines.
- symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; skin rash, itching or hives.
Do not use VFEND if you are taking any of the following medicines:
- pimozide (e.g., Orap), a medicine used to treat mental illness.
- quinidine (e.g., Kinidin Durules), a medicine used to treat irregular heartbeat.
- ivabradine (e.g., Coralan), a medicine used to treat heart problems.
- rifampicin (e.g., Rifadin, Rimycin), a medicine used to treat tuberculosis and other infections.
- carbamazepine (e.g., Tegretol, Teril), a medicine used to treat seizures.
- long-acting barbiturates such as phenobarbitone, medicines used to treat severe insomnia and seizures.
- rifabutin (e.g., Mycobutin) an antibiotic.
- ergotamine (e.g., Cafergot) or dihydroergotamine (e.g., Dihydergot), medicines used to treat migraine.
- sirolimus (e.g., Rapamune), a medicine used in transplant patients.
- efavirenz (e.g., Stocrin) (a medicine used to treat HIV infection) in doses of 400 mg or more once a day.
- ritonavir (e.g., Norvir, Kaletra) (a medicine used to treat HIV infection) in doses of 400 mg or more twice a day.
- St John's Wort (a herbal medicine).
- naloxegol, (e.g., Movantik), a medicine used to treat constipation caused by opioids (narcotic pain medicines).
- tolvaptan (e.g., Jinarc, Samsca) used to treat low levels of sodium in your blood or for kidney problems).
- venetoclax (e.g., Venclexta), a medicine used to treat blood cancers.
- lurasidone (e.g., Latuda), a medicine to treat schizophrenia and bipolar disorder.
VFEND should not be given to a child under the age of 2 years. Safety and effectiveness in children younger than 2 years has not been established.
Do not use this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor first.
Before you start to use it
Tell your doctor if you have allergies to foods, preservatives or dyes or any other medicines, especially antifungal medicines such as itraconazole (Sporanox), fluconazole (Diflucan), posaconazole (Noxafil) or ketoconazole (Nizoral) (not all brands given).
Tell your doctor if you have or have had any of the following medical conditions:
- heart problems.
- any problems affecting your kidneys.
- any problems affecting your liver. If you have liver disease your doctor may prescribe a lower dose.
- recent chemotherapy or stem cell transplant.
Tell your doctor if you are pregnant or plan to become pregnant. VFEND should not be used during pregnancy, unless indicated by your doctor. Effective contraception should be used in women of childbearing potential. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you are breast-feeding. VFEND should not be used whilst breastfeeding, unless indicated by your doctor. It is not known if the active ingredient voriconazole passes into breast milk. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell your doctor before you start taking VFEND.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines should not be taken with VFEND. These include (not all brands given):
- pimozide (e.g., Orap), a medicine used to treat mental illness.
- quinidine (e.g., Kinidin Durules), a medicine for irregular heartbeat.
- ivabradine (e.g., Coralan), a medicine used to treat heart problems.
- rifampicin (e.g., Rifadin, Rimycin), a medicine used to treat tuberculosis and other infections.
- carbamazepine (e.g., Tegretol, Teril), a medicine used to treat seizures.
- long-acting barbiturates such as phenobarbitone, medicines used to treat severe insomnia and seizures.
- rifabutin (e.g., Mycobutin) an antibiotic.
- ergotamine (e.g., Cafergot) or dihydroergotamine (e.g., Dihydergot), medicines used to treat migraine.
- sirolimus (e.g., Rapamune) a medicine used in transplant patients.
- efavirenz (Stocrin) (a medicine used to treat HIV infection) in doses of 400 mg or more once a day.
- ritonavir (e.g., Norvir, Kaletra) (a medicine used to treat HIV infection) in doses of 400 mg or more twice a day.
- St John's Wort, (a herbal medicine).
- venetoclax (e.g., Venclexta), a medicine used to treat blood cancers.
- naloxegol, (e.g., Movantik), a medicine used to treat constipation caused by opioids (narcotic pain medicines).
- tolvaptan (e.g., Jinarc, Samsca) used to treat low levels of sodium in your blood or for kidney problems).
- lurasidone (e.g., Latuda), a medicine to treat schizophrenia and bipolar disorder.
Some medicines and VFEND may interfere with each other. These include (not all brands given):
- efavirenz (Stocrin) (a medicine used to treat HIV infection) in doses below 400 mg once a day.
- ritonavir (Norvir, Kaletra) (a medicine used to treat HIV infection) in doses of 100 mg twice a day.
- warfarin (e.g., Marevan, Coumadin), a medicine used to stop blood clots.
- everolimus (e.g., Afinitor, Certican), a medicine used to treat cancer.
- other cancer drugs such as glasdegib, axitinib, bosutinib, cabozantinib, ceritinib, cobimetinib, dabrafenib, dasatinib, nilotinib, sunitinib, ibrutinib, ribociclib.
- tretinoin, a medicine used for the treatment of acne and acute promyelocytic leukemia.
- ivacaftor, (e.g., Kalydeco, Orkambi, Symdeko), a medicine used to treat cystic fibrosis.
- fluconazole (e.g., Diflucan), a medicine used to treat fungal infections.
- phenytoin (e.g., Dilantin), a medicine used to treat epilepsy.
- ciclosporin (e.g., Sandimmun, Neoral), a medicine used to prevent organ transplant rejection or to treat certain problems with the immune system.
- sulphonylureas, medicines used to treat diabetes such as glibenclamide, gliclazide and glipizide (e.g., Daonil, Diamicron, Minidiab).
- some antihistamines, medicines used to treat hayfever, allergic skin reactions, itching.
- theophylline (e.g., Nuelin), a medicine used to treat asthma.
- benzodiazepines (e.g., Valium), medicines used to treat insomnia or anxiety.
- eszopiclone, a medicine used to treat insomnia.
- statins (e.g., Zocor, Lipitor, Crestor), medicines used for lowering cholesterol.
- tacrolimus (e.g., Prograf), a medicine used in patients who have had a liver or kidney transplant.
- indinavir (e.g., Crixivan) and some other medicines used to treat HIV infection.
- omeprazole (e.g., Losec), a medicine used to treat indigestion, reflux and stomach or duodenal ulcers.
- methadone (used to treat heroin addiction).
- oral contraceptives (the Pill).
- vincristine, vinblastine or vinorelbine, medicines used in treating cancer (e.g., Vepesid).
- strong pain killers such as alfentanil (e.g., Rapifen), fentanyl (e.g., Durogesic, Actiq, Sublimaze) and oxycodone (e.g., Endone, Proladone).
- non-steroidal anti-inflammatory drugs, medicines used to treat pain and inflammation such as ibuprofen and diclofenac (e.g., Nurofen, Advil, Voltaren).
- letermovir (e.g., Prevymis) used to prevent viral infections after bone marrow transplant).
These medicines may be affected by VFEND or may affect how well it works. You may need different amounts of your medicines or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to use VFEND
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to use
Your doctor will tell you how much to use depending on your weight.
Adults
Treatment of invasive fungal infections
The usual dose of VFEND Tablets in adults weighing 40 kg and greater is 400 mg (two 200 mg tablets twice a day) for the first day and then 200 to 300 mg twice a day thereafter.
The usual dose of VFEND Oral Suspension in adults weighing 40 kg and greater is 10 mL twice a day for the first day and then 5 mL twice a day.
In adults weighing less than 40 kg the dose of VFEND Tablets and Oral Suspension is halved.
The usual dose of VFEND Powder for Injection is 6 mg/kg every 12 hours for the first day. The dose is then adjusted to 3 mg/kg or 4 mg/kg every 12 hours, depending on the type of infection you have.
In adults weighing less than 40 kg the dose of VFEND Tablets and Oral Suspension is halved.
Children
VFEND should not be given to a child under the age of 2 years.
Your doctor will determine the dose of VFEND required for your child.
Depending on how serious the infection is and how your child reacts to the medicine, your doctor may increase or decrease the dose.
Adolescents (12-16 years of age)
Adolescents aged 12-16 years of age are usually given the same dose as adults.
How to take it
VFEND Tablets
Swallow the tablets whole with a full glass of water.
VFEND Oral Suspension
Your pharmacist should advise you how to measure VFEND Oral Suspension using the multi-dosing syringe provided in the pack.
VFEND Oral Suspension should not be mixed with any other medication.
Please see instructions below before using VFEND Oral Suspension.
- Shake the bottle well before use.
- Remove cap and insert the tip of the syringe into the adaptor while the bottle is upright.
- Turn the bottle upside down while holding the syringe in place. Slowly pull back the plunger of the syringe until the required amount of VFEND Oral Suspension is withdrawn from the bottle.
- If large bubbles can be seen, slowly push the plunger back into the syringe. This will force the medicine back into the bottle. Repeat step 3 again.
- Turn the bottle back upright and then remove the syringe.
- Put the tip of the syringe into the mouth and point towards the inside of the cheek. SLOWLY push down the plunger of the syringe. Do not squirt the suspension out quickly.
If the medicine is to be given to a child, make sure the child is sitting or is held upright.
- Replace the bottle cap.
Cleaning and storing the syringe:
- The syringe should be rinsed after each dose. Pull the plunger out of the syringe and wash both parts by holding under warm running water.
- Dry the two parts. Push the plunger back into the syringe. Keep it in a clean safe place with the medicine.
VFEND Powder for Injection
VFEND Powder for Injection is given as an intravenous infusion by a doctor or trained nurse.
VFEND Powder for Injection is a powder which is mixed with Water for Injections and then diluted by your pharmacist or doctor. It is then given as an intravenous infusion into a vein.
When to take it
VFEND Tablets and Oral Suspension
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
Take your medicine 1 hour before food after a meal.
How long to take it
The length of time you take VFEND will depend on the type of infection you have.
If you have a weakened immune system or a difficult infection, you may need long-term treatment to prevent the infection from returning.
You may be switched from the intravenous infusion to VFEND Tablets or Oral Suspension once your condition improves.
Continue taking VFEND for as long as your doctor or pharmacist recommends. Do not stop taking VFEND because you are feeling better. If you do not complete the full course prescribed by your doctor, the infection may not clear completely or your symptoms may return.
If you forget to take it
If you forget to take one dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
VFEND Powder for injection will be given to you under close medical supervision. It is unlikely that a dose would be missed.
However, tell your doctor or pharmacist if you think that a dose has been forgotten.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (13 11 26) for advice or go to Accident and Emergency at your nearest hospital, if you think you or anyone else may have taken too much VFEND. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include upset stomach, diarrhoea, headache and sensitivity to light.
While you are using VFEND
Things you must do
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking VFEND. It may affect other medicines used during surgery.
Tell your doctor immediately if you develop a rash or blisters while taking VFEND. If this rash worsens, VFEND may need to be stopped.
Avoid going out in the sun for long periods of time while you are taking VFEND. VFEND can cause sensitivity to sunlight.
Tell your doctor if you notice any changes to your skin while you are taking VFEND.
Tell your doctor if you suffer from chronic, or long-lasting fatigue, muscle weakness, loss of appetite, weight loss or abdominal pain while on VFEND.
If the symptoms of your infection do not improve within a few days, or if they become worse, tell your doctor.
Make sure you follow your doctor's instructions and keep all appointments, including blood tests.
Your doctor should monitor the function of your liver and kidneys using blood tests. If you have liver disease, your doctor might lower your dose of VFEND or stop your VFEND treatment. Your doctor might also monitor the function of your pancreas.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are a woman of child-bearing age, you should avoid becoming pregnant while taking VFEND. If you become pregnant while taking VFEND, tell your doctor immediately.
Things you must not do
Do not use VFEND to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else even if they have the same condition as you or if their symptoms seem similar to yours.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you do not complete the full course prescribed by your doctor, the infection may not clear completely or your symptoms may return.
Things to be careful of
Be careful driving or operating machinery until you know how VFEND affects you.
You may experience changes to your vision, such as blurriness, colour changes or uncomfortable sensitivity to light.
If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous. Do not drive at night.
Children should be careful when riding bicycles or climbing.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking VFEND.
This medicine helps most people with fungal infections, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. If they occur, most are likely to be minor and temporary. However, some may be serious and need medical attention.
Do not be alarmed by the following lists of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- changes to your vision, such as blurred vision, colour changes or sensitivity to light
- irregular heartbeat
- nausea or feeling sick, vomiting
- headache
- stomach pain, indigestion, diarrhoea
- back pain in middle or upper back
- swelling of the arms or legs
- rash
- changes to your skin, such as skin eruptions or small lumps on the skin
- soreness at the injection site.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- swelling of the face, lips or tongue which may cause difficulty in swallowing or breathing
- asthma, wheezing, shortness of breath
- sudden or severe itching, skin rash, hives or blisters
- fainting, seizures or fits
- severe skin reaction such as painful red areas, large blisters, flaking of your skin, and bleeding in the lips, eyes, mouth, nose and genitals.
- yellowing of the skin or eyes, also called jaundice
- signs of frequent or worsening infections such as fever, severe chills, sore throat or mouth ulcers
- blood in urine
- signs of kidney failure such as tiredness, lack of appetite and reduced or greatly increased amount of urine
- convulsions, fits.
These may be signs of a serious allergic reaction or side effect. You may need urgent medical attention or hospitalisation. These side effects are rare.
Tell your doctor if you notice any other side effects.
Other side effects not listed above may also occur in some people.
After using VFEND
Storage
VFEND Tablets
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the blister pack, they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
VFEND Oral Suspension
Your pharmacist will store VFEND Oral Suspension in a powder form at 2°C to 8°C in the refrigerator before reconstitution.
Store prepared suspension in a cool dry place where the temperature stays below 30°C.
Discard suspension 14 days after preparation.
Do not store VFEND or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep your medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
VFEND Powder for Injection
VFEND Powder for Injection will be stored in the pharmacy or on the hospital ward and is kept below 30°C.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
VFEND Tablets
VFEND 50 mg Tablets are white, round tablets marked VOR50 on one side and Pfizer on the other.
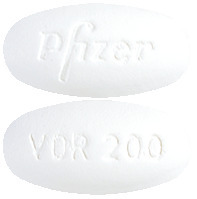
VFEND 200 mg Tablets are white, capsule shaped tablets marked VOR200 on one side and Pfizer on the other.
VFEND Oral Suspension
Once reconstituted, VFEND Oral Suspension is a white to off-white orange flavoured suspension. Each bottle contains 70 mL of suspension.
comes as a white powder in a clear, colourless, glass vial which contains 200 mg of voriconazole.
Ingredients
Active Ingredients
VFEND 50 mg Tablets contain 50 mg of voriconazole as the active ingredient.
VFEND 200 mg Tablets contain 200 mg of voriconazole as the active ingredient.
VFEND Oral Suspension contains 40 mg/mL of voriconazole as the active ingredient.
VFEND Powder for Injection contains 200 mg of voriconazole as the active ingredient in each 30 mL vial.
Inactive Ingredients
The 50 mg and 200 mg tablets contain the following other ingredients:
- lactose monohydrate
- maize starch (pregelatinised)
- croscarmellose sodium
- povidone
- magnesium stearate
- hypromellose
- titanium dioxide
- triacetin.
VFEND Oral Suspension contains the following other ingredients:
- sucrose
- colloidal anhydrous silica
- titanium dioxide
- sodium citrate dihydrate
- xanthan gum
- sodium benzoate
- citric acid
- natural orange flavour.
VFEND Powder for Injection contains the following other ingredients:
- water for injections
- sulfobutyl betadex sodium (SBECD).
Supplier
Pfizer Australia Pty Ltd
Sydney, NSW
Toll Free Number: 1800 675 229.
www.pfizermedinfo.com.au
Australian Registration Numbers
VFEND 50 mg blister pack
AUST R 82507.
VFEND 200 mg blister pack
AUST R 82505.
VFEND Powder for Oral Suspension
AUST R 99016.
VFEND Powder for Injection
AUST R 82503.
® = Registered Trademark.
© Copyright
This leaflet was prepared in November 2021.
Published by MIMS January 2022
 Dosage recommendations for other indications are provided in Tables 2 and 3.
Dosage recommendations for other indications are provided in Tables 2 and 3.






 The results of this comparative trial confirmed the results of an earlier trial in the primary treatment of patients with acute invasive aspergillosis (study 304). In this study, an overall success rate of 54% was seen in patients treated with voriconazole.
The results of this comparative trial confirmed the results of an earlier trial in the primary treatment of patients with acute invasive aspergillosis (study 304). In this study, an overall success rate of 54% was seen in patients treated with voriconazole.

 Voriconazole is designated chemically as (2R, 3S)-2-(2,4-difluorophenyl)-3-(5-fluoro-4-pyrimidinyl)-1-(1H-1,2,4-triazol-1-yl)-2-butanol with an empirical formula of C16H14F3N5O and a molecular weight of 349.3.
Voriconazole is designated chemically as (2R, 3S)-2-(2,4-difluorophenyl)-3-(5-fluoro-4-pyrimidinyl)-1-(1H-1,2,4-triazol-1-yl)-2-butanol with an empirical formula of C16H14F3N5O and a molecular weight of 349.3.