

What is in this leaflet
This leaflet answers some common questions about Vitrakvi. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Vitrakvi against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
What VITRAKVI is used for
Vitrakvi contains the active substance larotrectinib. It is used to treat a type of cancer called “Tyrosine Receptor Kinase (TRK) fusion cancer”. These cancers can appear in many different parts of the body.
Vitrakvi is used in adults and babies from 1 month old.
TRK fusion cancers always have a change in a gene called “Neurotrophic Tyrosine Receptor Kinase” (NTRK). The alteration in this gene causes the body to make a protein called “TRK fusion”. TRK fusion protein can cause uncontrolled cell growth and cancer.
Vitrakvi stops the TRK fusion proteins working and may slow or stop the cancer growing. It may also help to shrink the cancer.
Ask your doctor if you have any questions on how Vitrakvi works, or why this medicine has been prescribed for you.
This medicine is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine for children under the age of 1 month.
Before you take VITRAKVI
When you must not take it
Do not take Vitrakvi if:
- you have an allergy to larotrectinib (the active ingredient) or any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you are pregnant or plan to become pregnant or are beast-feeding.
If you are not sure whether you should start using this medicine, talk to your Doctor or health care professional.
If you have not told your doctor about any of the above, tell your doctor or health care professional before you start taking Vitrakvi.
Taking other medicines
Tell your doctor or pharmacist if you are taking, have recently taken or might take any other medicines including any that you get without a prescription from your pharmacy, supermarket, naturopath or health food shop such as vitamins, dietary supplements or herbal medicines.
Some medicines may affect the way Vitrakvi works or Vitrakvi may affect how other medicines work and cause serious side effects. In particular, tell your doctor if you are taking anything in this list or any other medicines:
- Medicines that may increase the amount of larotrectinib in your blood and increase the risk of side effect such as:
- itraconazole, ketoconazole, posaconazole and voriconazole, clarithromycin, telithromycin - used to treat fungal and bacterial infections
- atazanavir, indinavir, nelfinavir, ritonavir, saquinavir - used to treat HIV infection
- nefadozone - used to treat depression - Medicines that may decrease the amount of larotrectinib in your blood and make Vitrakvi less effective such as:
- phenytoin, carbamazepine and phenobarbital - medicines used to treat seizures
- St. John’s wort - herbal medicine used to treat depression
- Rifampicin - used to treat bacterial infections - Medicines which larotrectinib may decrease the amount of how much of them is found in the blood and make them less effective, such as:
- alfentanil - used as a narcotic pain medication
- cyclosporine, sirolimus, tacrolimus - used to prevent organ rejection in patients after transplantation
- Quinidine - used to treat abnormal heart rhythms
- dihydroergotamine, ergotamine - used in migraine or cluster headache attack
- fentanyl - used to treat chronic pain
- pimozide - antipsychotic drug used to control motor or verbal tics
These medicines may be affected by Vitrakvi or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take VITRAKVI
Before starting the therapy, a test will be used to determine whether you have NRTK gene alteration.
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
Always take this medicine exactly as your doctor or pharmacist has told you. It is important to do this as long as your doctor tells you. Check with your doctor, pharmacist, or nurse if you are not sure.
If you do not understand the instructions on the box or bottle, ask your doctor or pharmacist for help.
How much to take
Adults (from 18 years):
- the recommended dose of Vitrakvi in adults is 100 mg, two times a day (=200 mg total per day).
- You will either take 1 capsule of 100 mg or 4 capsules of 25 mg to make a total dose of 100 mg, two times a day.
- Your doctor will review your dose and change it as needed.
Babies and children (from 1 month old up to 18 years):
- Your child’s doctor will work out the right dose for your child.
- The maximum recommended dose is 100 mg (maximum of 1 capsule of 100 mg per dose, or 4 capsules of 25 mg per dose, or 5mL of the 20mg/mL solution), two times a day (=200 mg total per day).
- Your child’s doctor will review your dose and change it as needed.
How to take it
Swallow Vitrakvi capsules whole with a full glass of water.
Do not open, chew or crush the capsule.
Vitrakvi can be taken with or without food.
Do not eat grapefruit or drink grapefruit juice while taking Vitrakvi. This may increase the amount of Vitrakvi in your body.
If you or your child vomit (are sick) after taking Vitrakvi:
Do not take an extra dose, just take your next dose at the usual time.
For oral solution you need a bottle adapter (28 mm diameter) and a syringe that can be used to give medicines by mouth. Use a 1 mL syringe with 0.1 mL marks for doses less than 1 mL. Use a 5 mL syringe with 0.2 mL marks for doses of 1 mL or more.
- Press the bottle cap and turn it anti- clockwise to open the bottle.
- Put the bottle adapter into the bottle neck and make sure it is well fixed.
- Push the plunger fully into the syringe and then put the syringe in the adapter opening. Turn the bottle upside down.
- Fill the syringe with small amount of solution by pulling the plunger down, then push the plunger upwards to remove any large bubbles that are in the syringe.
- Pull the plunger down to the mark equal to the dose in mL prescribed by your doctor.
- Turn the bottle the right way up and take the syringe out of the adapter.
- Put the syringe in the mouth, pointing towards the inside of the cheek – this will help you swallow the medicine naturally. Slowly press the plunger in.
- Put the bottle cap on and tightly close the bottle - leave the adapter in the bottle.
If necessary, Vitrakvi may be administered via a nasogastric feeding tube. For details how to do so, please ask your doctor, pharmacist or nurse.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
If you or your child vomit (are sick) after taking Vitrakvi, do not take an extra dose; just take your next dose at the usual time.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
If you stop taking Vitrakvi
Do not stop taking this medicine without talking to your doctor first. It is important to take Vitrakvi for as long as your doctor prescribes it for you.
If you are not able to take the medicine as your doctor prescribed, or you feel you do not need it anymore, contact your doctor right away.
If you have further questions on the use of this medicine, ask your doctor, pharmacist or nurse.
If you forget to take it
Do not take a double dose to make up for the dose that you missed.
Take your next dose at the usual time
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
If you think that you or anyone else may have taken too much Vitrakvi, immediately contact your doctor, pharmacist, nurse or the Poisons Information Centre for advice, or go to Accident and Emergency at the nearest hospital. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
You can contact the Poisons Information Centre by dialling:
- Australia: 13 11 26
- New Zealand: 0800 POISON or 0800 764 766.
Take the medicine package and this leaflet with you. You may be at a higher risk of experiencing the side effects described in Side Effects section.
While you are using VITRAKVI
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Vitrakvi.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
Tell your doctor, pharmacist or nurse straight away if you get any of these symptoms during treatment:
nervous system problems such as feeling dizzy, difficulty walking normally, tingling, feeling numb, or a burning feeling in your hands and feet.
Vitrakvi can increase the amount of some substances in the blood which are made by the liver. Your doctor will do blood tests before and during treatment to check the level of these substances and check how well your liver is working.
Your doctor may reduce the dose of Vitrakvi and may delay or stop Vitrakvi treatment permanently (see also section ‘Side Effects’).
If you become pregnant while taking this medicine, tell your doctor immediately.
- Avoid becoming pregnant during treatment with Vitrakvi. Based on how Vitrakvi works, the risk of permanent harm or birth defects to the baby cannot be ruled out.
- If you are able to become pregnant, your doctor may do a pregnancy test before you start treatment with Vitrakvi.
- You should also use effective methods of contraception while taking Vitrakvi and for at least one month after the last dose of Vitrakvi. Ask your doctor about the best method of contraception for you. Tell your doctor if you think you are pregnant or plan on becoming pregnant.
- If you become pregnant during treatment with Vitrakvi or in the first month after your last dose, tell your doctor straight away.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take Vitrakvi to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not give this medicine to infants under 1 month of age as it is not known if Vitrakvi is safe and effective in these infants.
Do not breast-feed while taking this medicine and for 3 days after the last dose of Vitrakvi. There are no data on the presence of larotrectinib in human milk, the effects of larotrectinib on the breastfed child, or the effects of larotrectinib on milk production.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects. If possible, your doctor will gradually reduce the amount you take each day before stopping the medicine completely.
Things to be careful of
Be careful driving or operating machinery until you know how Vitrakvi affects you. This medicine may cause dizziness, tiredness and nausea in some people. If you have any of these symptoms, do not drive, cycle, use any tools or machines or do anything else that could be dangerous. Children should be careful when riding bicycles or climbing trees.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Vitrakvi.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor as soon as possible if you notice any of the following:
- Difficulty walking normally (gait disturbance)
- Abnormal sense of touch, tingling (paraesthesia)
- Change in how things taste (dysgeusia)
- Weight increase
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- Dizziness
- Feeling tired (fatigue)
- Stomach upset, including feeling sick (nausea), being sick (vomiting) or constipation
- Muscle pain (myalgia)
- Muscle weakness
- Reduction in the number of red blood cells which makes you feeling tired, looking pale and you may feel your heart pumping (anaemia)
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
Some of these side effects can only be found when your doctor does tests from time to time to check your progress. These includes:
- Increased liver enzymes shown in blood tests (called “alanine aminotransferase” and “aspartate aminotransferase”)
- Decrease number of white blood cells (called “neutrophils” and “leukocytes”)
- Abnormal results of tests to check for liver disease or bone disorders (high levels of “alkaline phosphatase”)
Your doctor will tell you if there are any changes in your blood test that might need treatment.
After using VITRAKVI
Storage
For capsules, store below 25°C (77°F).
For solution, keep refrigerated at 2°C to 8°C (36°F to 46°F). Do not freeze.
Do not store Vitrakvi or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Store in the original package to protect it from moisture.
Keep the bottle tightly closed.
Do not use this medicine if you notice that capsules look damaged.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Vitrakvi comes in two forms; capsules and solution.
- Vitrakvi 25 mg hard capsules are white opaque hard gelatin capsule, size 2, with blue printing of “BAYER” cross and “25 mg” on body of capsule
- Vitrakvi 100 mg hard capsules are white opaque hard gelatin capsule, size 0, with blue printing of “BAYER” cross and “100 mg” on body of capsule
- Vitrakvi 20 mg/mL oral solution are 100 mL clear yellow to orange liquid solution
Ingredients
The active ingredient in Vitrakvi is larotrectinib; it comes in capsules and solution forms.
For capsule
Active ingredient:
larotrectinib
Inactive ingredients:
- Gelatin
- Titanium dioxide
- Printing ink –blue: Shellac, FD&C Blue #2 aluminium lake, titanium dioxide, propylene glycol, ammonia solution and dimethicone
For solution
Active ingredient:
larotrectinib
Inactive ingredients:
- Purified water
- Hydroxypropyl betadex
- Sodium citrate
- Ora-Sweet®: purified water, sucrose, glycerol, sorbitol, citric acid, sodium dihydrogen phosphate, flavouring and preservative agents - methylparahydroxybenzoate and potassium sorbate
- Natural Masking Type Flavor: glycerol, natural flavour ingredients
- Natural Bitterness Masking Type Flavor: glycerol, natural flavour ingredients
- Bitterness Masking Flavor: propylene glycol, natural flavour
- FONATECH® Taste Modifier Flavor: propylene glycol, glycerol, natural flavour
Supplier
Vitrakvi is supplied in Australia by:
Bayer Australia Ltd
ABN 22 000 138 714
875 Pacific Highway
Pymble NSW 2073
Australian registration number:
- Vitrakvi 25 mg hard capsules: AUST R 320237
- Vitrakvi 100 mg hard capsules: AUST R 320238
- Vitrakvi 20mg/mL oral solution: AUST R 320239
This leaflet was prepared in September 2020.
®Registered Trademark of the Bayer Group, Germany.
® Bayer Australia Ltd.
All rights reserved.

Published by MIMS March 2022

 Vitrakvi should be permanently discontinued in patients who are unable to tolerate Vitrakvi after three dose modifications.
Vitrakvi should be permanently discontinued in patients who are unable to tolerate Vitrakvi after three dose modifications.
 See Table 5.
See Table 5.

 For the pooled efficacy analysis, the primary efficacy endpoint was overall response rate (ORR), as determined by an Independent Review Committee (IRC). ORR was defined as the proportion of patients with the best overall response of confirmed complete response (CR) or confirmed partial response (PR) based on RECIST version 1.1 for solid tumors. Secondary efficacy endpoints for the pooled analysis included time to first response, duration of response and disease-control rate (DCR; best overall response of CR, PR, or stable disease lasting 16 or more weeks). Additional secondary efficacy endpoints were progression-free survival (PFS) and overall survival (OS) and time on treatment.
For the pooled efficacy analysis, the primary efficacy endpoint was overall response rate (ORR), as determined by an Independent Review Committee (IRC). ORR was defined as the proportion of patients with the best overall response of confirmed complete response (CR) or confirmed partial response (PR) based on RECIST version 1.1 for solid tumors. Secondary efficacy endpoints for the pooled analysis included time to first response, duration of response and disease-control rate (DCR; best overall response of CR, PR, or stable disease lasting 16 or more weeks). Additional secondary efficacy endpoints were progression-free survival (PFS) and overall survival (OS) and time on treatment. Responses were seen in patients regardless of their age, tumor type, NTRK gene involved or NTRK gene fusion partner. In the paediatric sub-population (n = 55), the ORR was 91%.
Responses were seen in patients regardless of their age, tumor type, NTRK gene involved or NTRK gene fusion partner. In the paediatric sub-population (n = 55), the ORR was 91%.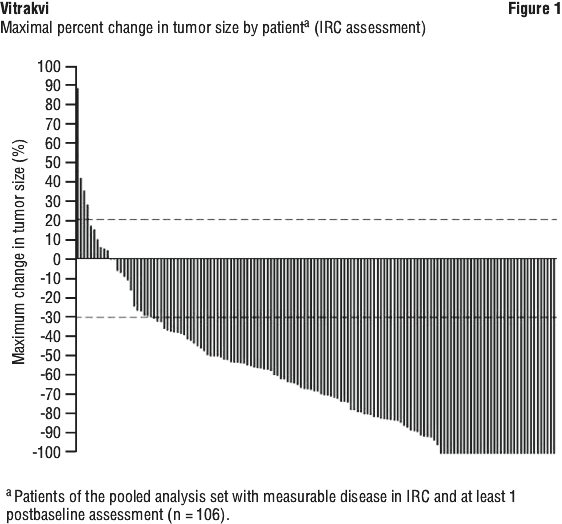

 Chemical name: (3S)-N-{5-[(2R)-2-(2,5-Difluorophenyl)-1-pyrrolidinyl] pyrazolo[1,5a]pyrimidin-3-yl}-3-hydroxy-1-pyrrolidinecarboxamide sulfate.
Chemical name: (3S)-N-{5-[(2R)-2-(2,5-Difluorophenyl)-1-pyrrolidinyl] pyrazolo[1,5a]pyrimidin-3-yl}-3-hydroxy-1-pyrrolidinecarboxamide sulfate.