

What is in this leaflet
Please read this leaflet carefully before you start taking VOLIBRIS. This leaflet answers some common questions about VOLIBRIS. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking VOLIBRIS against the benefits they expect it will have for you.
If you have any questions about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What VOLIBRIS is used for
VOLIBRIS contains the active ingredient ambrisentan which is a type of medicine called an endothelin receptor antagonist (ERA). It is used to treat adults with pulmonary arterial hypertension (PAH), which is high blood pressure in the blood vessels (the pulmonary arteries) that carry blood from the heart to the lungs. In people with PAH, these arteries get narrower, so the heart has to work harder to pump blood through them. This causes people to become tired, dizzy and short of breath. VOLIBRIS widens the pulmonary arteries, making it easier for your heart to pump blood through them. This lowers the blood pressure and relieves your symptoms.
VOLIBRIS is not recommended for use in children as there have been no studies of its effects in children.
VOLIBRIS must not be used for the treatment of idiopathic pulmonary fibrosis (IPF) and must not be used in patients who have IPF with or without secondary pulmonary hypertension.
VOLIBRIS is available as 5 mg and 10 mg tablets.
This medicine is available only with a doctor's prescription.
Before you take VOLIBRIS
Do not take VOLIBRIS if you have an allergy to:
- any medicine containing ambrisentan
- any of the ingredients listed at the end of this leaflet
- any other similar medicine.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take VOLIBRIS if you are pregnant or planning to become pregnant. It may affect your developing baby if you take it during pregnancy or if you become pregnant soon after you stop treatment.
If it is possible you could become pregnant, you must use reliable birth control (contraception) while using VOLIBRIS and for 3 months after you stop taking it.
Use at least two reliable forms of birth control (contraception) while you are taking VOLIBRIS. Talk to your doctor about this.
If you are a woman who could become pregnant, your doctor will ask you to take a pregnancy test before you take VOLIBRIS.
If you are a male you should avoid exposing your partner to your semen by use of appropriate contraception e.g. condoms.
If you do become pregnant, talk to your doctor immediately.
Do not take VOLIBRIS if:
- you have scarring of the lungs of unknown cause (a condition known as idiopathic pulmonary fibrosis or IPF)
- you have or have had a serious liver problem
- you have raised levels of some liver enzymes (detected by blood tests).
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you are breast-feeding or plan to breast feed. Your doctor can discuss with you the risks and benefits involved.
Tell your doctor if you have or have had any of the following medical conditions:
- liver disease
- heart disease called right heart failure
- a low number of red blood cells (anaemia)
- if you have swelling, especially of the ankles and feet (oedema)
- low blood pressure
- kidney problems.
If any of these applies to you, your doctor will decide if VOLIBRIS is suitable for you.
Blood tests
Your doctor will take blood tests to check:
- whether you have a reduced number of red blood cells (anaemia)
- that your liver is working properly.
These blood tests will be taken before you start taking VOLIBRIS and throughout your treatment with VOLIBRIS.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and VOLIBRIS may interfere with each other. This includes:
- cyclosporin A (a medicine taken if you have an organ transplant). If you take cyclosporin A, do not take more than one 5 mg tablet of VOLIBRIS, once a day
- ketoconazole (medicine for fungal infection).
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take VOLIBRIS
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much to take
VOLIBRIS
The usual dose of VOLIBRIS is 5 mg, once a day. Your doctor may decide to increase your dose to 10 mg, once a day.
VOLIBRIS with Tadalafil
When used in combination with Tadalafil (another PAH medicine), VOLIBRIS is usually started at 5 mg daily, and should be increased to 10 mg once a day, if you can tolerate a higher dose.
If you take cyclosporin A, do not take more than one 5 mg tablet of VOLIBRIS, once a day.
How to take it
Swallow the tablet whole, with a glass of water.
VOLIBRIS tablets should not be split, crushed or chewed.
You can take VOLIBRIS with or without food.
VOLIBRIS tablets are packaged in a special child resistant blister pack.
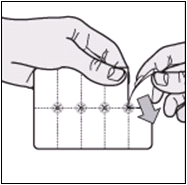
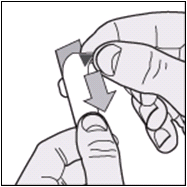
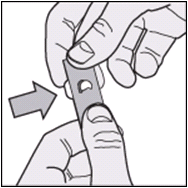
The diagrams below provide instructions on how best to remove the tablets from the packaging.
- Tear along the perforated lines to separate one "pocket" from the blister strip.
- Starting at the coloured corner, gently lift and peel back the outer layer over the pocket, exposing the foil beneath.
- Push out the tablet through the exposed foil layer.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it
Continue taking your medicine for as long as your doctor tells you. This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
Take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much VOLIBRIS
Immediately telephone your doctor or Poisons Information Centre (in Australia, telephone 131126) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much VOLIBRIS. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using VOLIBRIS
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking VOLIBRIS.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
You must use reliable birth control (contraception) while using VOLIBRIS and for 3 months after you stop taking it. Use at least two reliable forms of birth control (contraception).
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are a male, you should avoid exposing your partner to your semen by use of appropriate contraception e.g. condoms.
Keep all of your doctor's appointments so that your progress can be checked.
Your doctor will need to take regular blood tests while you are taking this medicine to ensure your liver function and red blood cell (anaemia) levels remain normal.
Things you must not do
Do not take VOLIBRIS to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
It is not known whether VOLIBRIS affects your ability to drive or use machines. However, the symptoms of your condition can make you less fit to drive.
Do not drive or operate machines if you're feeling unwell.
VOLIBRIS contains the colouring agent Allura red AC Aluminium Lake (FD&C Red #40), which may cause allergic-type reactions.
Check with your doctor that VOLIBRIS is suitable for you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking VOLIBRIS.
This medicine helps most people with pulmonary arterial hypertension, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Very common side effects (could affect more than one in 10 people) include:
- swelling (oedema), especially in the ankles and feet
- headache
- a runny or blocked nose, congestion or pain in the sinuses
- dizziness
- palpitations (fast or irregular heart beat)
- feeling tired, lack of energy (fatigue)
- worsening shortness of breath shortly after starting VOLIBRIS
- signs of anaemia, such as tiredness, weakness, shortness of breath and feeling generally unwell
- when taking VOLIBRIS in combination with Tadalafil (another PAH) medicine. All the above apply plus:
- vomiting
- flushing (redness of the skin)
- chest pain/discomfort
- nausea
- diarrhoea.
Other common side effects (could affect up to one in 10 people) include:
- constipation
- pain in your stomach
- feeling weak
- vomiting
- heart failure
- hypotension
- fainting
- abnormal blood tests for liver function
- runny nose
- chest pain or discomfort
- flushing
- nose bleed
- rash
- tinnitus (ringing or buzzing in the ear) when taking VOLIBRIS in combination with Tadalafil
- allergic reactions: you may notice a rash or itching or swelling (usually of the face, lips or throat), which may cause difficulty in breathing or swallowing
- visual disturbance (blurred vision or changes in your ability to see clearly).
Uncommon side effects include:
- signs that your liver may not be working as it normally should include:
- loss of appetite
- sick (nausea)
- vomiting
- fever
- unusual tiredness
- pain in the stomach (abdominal pain)
- yellow colouring of your skin or the whites of your eyes (jaundice)
- your urine turns dark in colour
- itching of the skin
- abnormal liver function which may show up in your blood tests - sudden hearing loss, in combination with Tadalafil.
If you notice any of these signs, tell your doctor immediately.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After using VOLIBRIS
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store VOLIBRIS or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What VOLIBRIS looks like
VOLIBRIS is presented in packs of 30 tablets.
VOLIBRIS is available in two strengths - 5 mg and 10 mg.
The 5 mg tablet is a pale pink, square convex tablet engraved with 'GS' on one face and 'K2C' on the other.
The 10 mg tablet is a deep pink, oval convex tablet engraved with 'GS' on one face and 'KE3' on the other.
Ingredients
VOLIBRIS contains the active ingredient ambrisentan.
VOLIBRIS also contains:
- Microcrystalline cellulose,
- Lactose monohydrate,
- Croscarmellose sodium,
- Magnesium stearate,
- Polyvinyl alcohol,
- Purified talc,
- Titanium dioxide,
- Macrogol 3350 (PEG 3350),
- Lecithin (Soya) USNF (E322),
- Allura Red AC Aluminium Lake (FD&C Red #40). See Section ‘While you are using VOLIBRIS’ for further information about this ingredient.
Distributor
VOLIBRIS is supplied in Australia by:
GlaxoSmithKline Australia Pty Ltd
Level 4, 436 Johnston Street
Abbotsford, Victoria, 3067
Australia
Where to go for further information:
This leaflet was prepared on 01 December 2020.
The information provided applies only to VOLIBRIS.
VOLIBRIS:
5 mg - AUST R 143743
10 mg - AUST R 143739
Version 15
VOLIBRIS is a registered trade mark of Gilead. Used under licence by the GSK group of companies.
© 2020 GSK group of companies or its licensor.
Published by MIMS April 2021


 Results from the extension studies also indicates that the benefits were maintained at 48 weeks. The mean change in 6MWD from baseline at week 48 was +35.2 m (95% CI: 13.0 to 57.5; n = 68) for the 5 mg dose, and +30.2 m (95% CI: 10.8 to 49.6; n = 32) for the 10 mg dose.
Results from the extension studies also indicates that the benefits were maintained at 48 weeks. The mean change in 6MWD from baseline at week 48 was +35.2 m (95% CI: 13.0 to 57.5; n = 68) for the 5 mg dose, and +30.2 m (95% CI: 10.8 to 49.6; n = 32) for the 10 mg dose.



 The results for analyses of time to adjudicated clinical failure and to the first of each component of clinical failure are shown in Figure 2.
The results for analyses of time to adjudicated clinical failure and to the first of each component of clinical failure are shown in Figure 2. Efficacy of first line treatment with ambrisentan + tadalafil on time to clinical failure was seen across subgroups (see Figure 3).
Efficacy of first line treatment with ambrisentan + tadalafil on time to clinical failure was seen across subgroups (see Figure 3).

 In patients with PAH who received first line combination therapy with ambrisentan and tadalafil, a greater decrease from baseline in NT-pro BNP relative to pooled monotherapy was observed (geometric least squares mean percent decreases of 67% versus 50%, respectively). Similar results were observed for the comparisons of combination therapy versus ambrisentan monotherapy group (56% decrease) and tadalafil monotherapy group (44% decrease). The decrease in NT-pro BNP was observed early (week 4) and was sustained through week 24.
In patients with PAH who received first line combination therapy with ambrisentan and tadalafil, a greater decrease from baseline in NT-pro BNP relative to pooled monotherapy was observed (geometric least squares mean percent decreases of 67% versus 50%, respectively). Similar results were observed for the comparisons of combination therapy versus ambrisentan monotherapy group (56% decrease) and tadalafil monotherapy group (44% decrease). The decrease in NT-pro BNP was observed early (week 4) and was sustained through week 24.
