

What is in this leaflet
This leaflet answers some common questions about VOLIROP.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking VOLIROP against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What VOLIROP used for
VOLIROP contains the active ingredient carvedilol.
VOLIROP is used to treat
- heart failure,
- high blood pressure, which is called hypertension.
VOLIROP belongs to a group of medicines called beta-blockers. These medicines work by relaxing tightened blood vessels and slowing the heart rate. VOLIROP has the additional effect of being an antioxidant.
Heart Failure:
Heart failure occurs when the heart can no longer pump blood strongly enough for the body's needs. Often the heart grows in size to try to improve the blood flow but this can make the heart failure worse.
Symptoms of heart failure include shortness of breath and swelling of the feet or legs due to fluid build-up.
VOLIROP reduces the pressure that the heart has to pump against as well as controlling your heart rate. Over 6 months or more this will reduce the size of an oversized heart and increase its efficiency.
VOLIROP reduces the chances of you being admitted to hospital and/ or dying from this condition.
VOLIROP is often used with other medicines to treat heart failure.
Hypertension:
All people have blood pressure. This pressure helps to push blood all around your body. Your blood pressure changes during the day, depending on how busy you are or how you are feeling.
You have hypertension (high blood pressure) when your blood pressure stays higher than is needed, even when you are calm and relaxed.
Regular blood pressure checks are the only way of knowing that you have hypertension. There are usually no symptoms of hypertension and you may feel fine. If hypertension is not treated, serious health problems such as stroke, heart disease and kidney failure may occur.
VOLIROP helps to lower your blood pressure.
Your doctor, however, may have prescribed VOLIROP for another purpose.
Ask your doctor if you have any questions about why VOLIROP has been prescribed for you.
VOLIROP is not addictive.
This medicine is available only with a doctor's prescription.
Before you take VOLIROP
When you must not take VOLIROP
Do not take VOLIROP if:
- you have had an allergic reaction to:
- any medicine containing carvedilol
- any ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- you have asthma or other conditions which make you short of breath from time to time.
- you have a history of allergic problems, including hayfever. Symptoms of an allergy may include: rash, itching, watery eyes or sneezing
- you have a history of a very slow heart rate or uneven heart beating.
- you have certain other heart conditions.
- you have liver problems including liver failure.
- you have very low blood pressure.
- the package is torn or shows signs of tampering.
- the expiry date (EXP) printed on the pack has passed or if the tablets appear damaged in some way.
If you take this medicine after the expiry date has passed, it may not work as well.
If you are not sure if you should be taking VOLIROP, talk to your doctor.
Do not give VOLIROP to people under 18 years of age.
Safety and effectiveness in children have not been established.
Before you start to take VOLIROP
Tell your doctor if:
- you are pregnant or plan to become pregnant.
It is not known whether VOLIROP is harmful to an unborn baby when taken by a pregnant woman. Your doctor will discuss the risks and benefits of using VOLIROP during your pregnancy. - you are breastfeeding or plan to breastfeed.
VOLIROP passes into breast milk. Your doctor will discuss the risks and benefits of taking VOLIROP if you are breast-feeding. - you have any other health problems, especially the following:
- angina or chest pain/tightness which occurs even when you are at rest (also called unstable angina)
- low blood pressure
- high blood pressure which varies widely
- very poor circulation to your fingers and/or toes (also called peripheral vascular disease)
- a history of poor kidney function
- chronic bronchitis or emphysema causing breathing difficulties
- diabetes
- sudden low blood sugar levels (also called hypoglycaemia)
- thyroid disorders
- severe allergic reactions causing swelling and/or difficulty breathing
- a rare cancer of the adrenal gland called phaeochromocytoma which is not being treated with other medicines.
- skin disease such as psoriasis (hardened patches of red skin)
- you are allergic to any other medicines, foods, dyes or preservatives
- you plan to have surgery.
Your surgeon and anaesthetist should know well ahead of the date of your surgery so they can allow for your condition and medications.
If you have not told your doctor about any of the above, tell them before you start taking VOLIROP.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought from a pharmacy, supermarket or health food shop.
Some medicines may interfere with VOLIROP. These medicines include:
- rifampicin, a medicine used to treat tuberculosis (e.g. Rimycin®, Rifadin®)
- cimetidine, a medicine used to treat stomach ulcers or reflux (e.g. Tagamet®, Magicul®)
- digoxin, a medicine used to treat heart failure (e.g. Lanoxin®)
- monoamine-oxidase inhibitors (MAOIs) such as phenelzine (Nardil®) and tranylcypromine (Parnate®), medicines used to treat depression
- clonidine, a medicine used to treat high blood pressure, migraine or menopausal symptoms (e.g. Catapres®)
- diltiazem, a medicine used to treat high blood pressure or angina (e.g. Cardizem®, Coras®, Diltahexal®, Dilzem®, Vasocardol®)
- verapamil, a medicine used to treat high blood pressure, angina or fast heart rate (e.g. Isoptin®, Cordilox®, Anpec®)
- drugs for when your heart doesn't beat smoothly, including disopyramide (Rythmodan®), mexiletine (Mexitil®), lignocaine, flecainide (Tambocor®) andamiodarone (Cordarone®, Aratac®).
- drugs for diabetes, including insulin injections, glibenclamide (Daonil®, Glimel®), metformin (Diabex®, Diaformin®, Glucohexal®, Glucomet®, Glucophage®, gliclazide, (Diamicron®), glipizide (Minidiab®, Melizide®).
- cyclosporin, a medicine used to treat certain problems with the immune system (e.g. Neoral®, Cicloral®, Sandimmun®).
- Aspirin and other pain relievers or non-steroidal anti-inflammatory medicines such as ibuprofen (Nurofen®) or naproxen (Naprosyn®).
- medicines which may relieve asthma or help you breath better such as salbutamol (Ventolin®) and salmeterol (Serevent / Seretide®).
- fluoxetine, a medicine used to treat depression and other conditions (e.g. Prozac®)
- other medicines that may help lower your blood pressure.
These medicines may be affected by VOLIROP, or may affect how well it works. You may need to use different amounts of your medicine, or you may need to take different medicines. Your doctor will advise you.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking VOLIROP.
Ask your doctor or pharmacist if you are not sure about this list of medicines.
How to take VOLIROP
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
How much to take VOLIROP
Take VOLIROP exactly as your doctor has prescribed.
Your doctor will tell you how many VOLIROP to take each day. This depends on your condition and whether or not you are taking other medicines.
Heart Failure
The usual starting dose in heart failure is 3.125mg twice daily. The dose is usually increased every two weeks to 6.25mg twice daily, 12.5mg twice daily and then 25mg twice daily. However, this may be done more slowly if side effects occur. If the tablets slow your heart too much you may go back to a lower dose.
High Blood Pressure
- Adults:
The recommended dose for initiation of therapy is 12.5 mg a day for the first two days.
Thereafter, the recommended dosage is 25 mg once a day.
If necessary, the dosage may be increased every two weeks up to the recommended maximum daily dose of 50 mg given once a day or in divided doses (twice daily). - Elderly:
The recommended dose for initiation of therapy is 12.5 mg once daily, which has provided satisfactory control in some patients. If the response is inadequate, the dose may be increased every two weeks up to the recommended maximum daily dose.
Your doctor will monitor you carefully each time the dose is increased.
How to take VOLIROP
Swallow tablets whole or halved with a glass of water.
Do not crush or chew the tablets.
When to take VOLIROP
Take VOLIROP during or immediately after a meal, at about the same times each day. Taking your medicine at the same time each day will have the best effect. It will also help you to remember when to take it.
If you take VOLIROP on an empty stomach, it may increase the risk of some of the side effects.
How long to take VOLIROP
Continue taking VOLIROP until your doctor tells you to stop.
Make sure you have enough to last over weekends and holidays.
It is very important that VOLIROP is not ceased suddenly. If you are to stop taking VOLIROP your doctor will advise you to reduce the dose slowly over approximately two weeks.
If you forget to take VOLIROP
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering your dose, ask your pharmacist for some hints.
In case of an overdose
If you think you or anyone else may have taken too much VOLIROP, immediately telephone your doctor, or Poisons Information Centre (telephone 13 11 26), or go to Accident and Emergency at your nearest hospital. Do this even if there are no signs of discomfort or poisoning.
Keep telephone numbers for these places handy.
If you are not sure what to do, contact your doctor or pharmacist.
You may need urgent medical attention.
The following are some symptoms, which may or may not occur.
- low blood pressure, causing dizziness or fainting
- a very slow heart rate
- difficulty breathing
- vomiting
- shock
- seizures.
While you are taking VOLIROP
Things you must do
Tell all doctors, dentists and pharmacists who are treating you that you are taking VOLIROP. You should also tell your surgeon and anaesthetist if you are having surgery.
If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly.
Make sure you drink enough water during exercise and hot weather when you are taking VOLIROP, especially if you sweat a lot.
Tell your doctor if you become pregnant while taking VOLIROP.
Tell your doctor that you are taking VOLIROP if you are going to have any laboratory tests.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Tell your doctor if you feel the tablets are not helping your condition.
Be sure to keep all of your appointments with your doctor so that your progress can be checked. Your doctor may examine your eyes, and test your blood glucose and kidney function from time to time.
Things you must not do
Do not stop taking VOLIROP or change the dose without first checking with your doctor.
Do not let yourself run out of medicine over the weekend or on holidays. VOLIROP should only be stopped by gradually reducing the amount over a two-week period.
Do not give VOLIROP to anyone else even if they have the same condition as you.
Do not use VOLIROP to treat other complaints unless your doctor says to.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how VOLIROP affects you.
VOLIROP may affect your ability to drive a car or operate machinery when started or when the dosage is increased.
If you wear contact lenses you may also notice a reduction in the amount of tear fluid in your eyes.
When taken with grapefruit juice the amount of VOLIROP absorbed by your body may be increased.
Side Effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking VOLIROP. VOLIROP helps most people with heart failure but it may have unwanted side effects in a few people. All medicines can have side effects.
Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- headache (this is usually mild and happens at the start of your treatment)
- tiredness, drowsiness
- low blood pressure. The signs include feeling dizzy or lightheaded especially after you stand up
- abnormal or blurry vision
- slow heart rate
- diarrhoea
- nausea or vomiting
- bronchitis
- loss of control of blood sugar in diabetics
- weight increase
- fluid retention. The signs include overall swelling of parts of your body for example your hands, feet, ankles and legs.
- unusual hair loss or thinning
These are the more common side effects of VOLIROP. Mostly these are mild and will decrease as you get used to your medicine.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- itching, dark urine, loss of appetite, yellowing of skin or eyes, or feeling "flu-like" with no clear cause.
- shortness of breath or swelling of the mouth or tongue
- uneven heart beating
- swelling of the feet or legs due to fluid build up
- bleeding or bruising more easily than normal
These may be serious side effects. You may need urgent medical attention. Serious side effects are rare.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor or pharmacist if you don't understand anything in this list.
After Taking VOLIROP
Storage
Keep your tablets in the blister until it is time to take them. If you take the tablets out of the blister pack they may not keep well. VOLIROP is known to fade when exposed to light.
Keep VOLIROP in a cool dry place where the temperature stays below 25°C.
Do not store it, or any other medicine, in a bathroom or near a sink, or any other place where there is high humidity.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
Keep VOLIROP where young children cannot reach it. A locked cupboard at least one-and-a- half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking VOLIROP, or the tablets have passed their expiry date, ask your pharmacist what to do with any tablets that are left over.
Product Description
What it looks like
VOLIROP 3.125, 6.25, 12.5 & 25 (3.125, 6.25, 12.5 & 25 mg carvedilol)
are presented in pack size of 30 & 60 tablets in blister and 30, 60 & 1000 tablets in bottle.
- VOLIROP 3.125
(Blister pack: AUST R 174802 & Bottle: AUST R 174786)
White to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘01’ on the other side. - VOLIROP 6.25
(Blister pack: AUST R 174796 & Bottle: AUST R 174806)
White to off-white, oval shaped, film-coated tablets debossed with ‘F 57’ on one side and deep break line on the other side. - VOLIROP 12.5
(Blister pack: AUST R 174795 & Bottle: AUST R 174800)
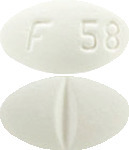
White to off-white, oval shaped, film-coated tablets debossed with ‘F 58’ on one side and deep break line on the other side. - VOLIROP 25
(Blister pack: AUST R 174788 & Bottle: AUST R 174789)
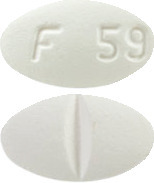
White to off-white, oval shaped, film-coated tablets debossed with ‘F 59’ on one side and deep break line on the other side.
Ingredients
Active Ingredients:
Each tablet may contain either 3.125mg, 6.25mg, 12.5mg or 25mg of carvedilol
Other Ingredients:
- Lactose
- Colloidal anhydrous silica
- Crospovidone (Type A & Type B)
- Povidone
- Sucrose
- Magnesium stearate
- Macrogol 400
- Polysorbate 80
- Titanium dioxide &
- Hypromellose
Please read this leaflet carefully before you start taking VOLIROP. You may wish to keep it to read again.
Name and Address of the Sponsor
Aurobindo Pharma Australia Pty Ltd
Unit 3 North Rydelink
277-283 Lane Cove Road
Macquarie Park NSW 2113
Australia
Date of Approval
7 November 2011
Published by MIMS August 2012


 The majority of patients were hospitalised for cardiovascular reasons. Treatment with carvedilol resulted in lower rates for almost all cardiac hospitalisations (worsening heart failure, atrial and ventricular and tachyarrhythmias, myocardial infarction and unstable angina pectoris). The number of patients hospitalised for symptomatic bradycardia and symptomatic heart block were slightly higher in the carvedilol treated patients than placebo, although the total number of patients hospitalised was low (1.3% and 0.8% respectively for bradycardia and 0.3% and 0.1% respectively for heart block). The number of patients hospitalised for noncardiovascular events was similar in both groups (placebo 11.2%, carvedilol 10.6%).
The majority of patients were hospitalised for cardiovascular reasons. Treatment with carvedilol resulted in lower rates for almost all cardiac hospitalisations (worsening heart failure, atrial and ventricular and tachyarrhythmias, myocardial infarction and unstable angina pectoris). The number of patients hospitalised for symptomatic bradycardia and symptomatic heart block were slightly higher in the carvedilol treated patients than placebo, although the total number of patients hospitalised was low (1.3% and 0.8% respectively for bradycardia and 0.3% and 0.1% respectively for heart block). The number of patients hospitalised for noncardiovascular events was similar in both groups (placebo 11.2%, carvedilol 10.6%). The following adverse events were reported more frequently with carvedilol in placebo controlled trials in patients with congestive heart failure.
The following adverse events were reported more frequently with carvedilol in placebo controlled trials in patients with congestive heart failure.