

What is in this leaflet
Please read this leaflet carefully before you take VOTRIENT.
This leaflet answers some common questions about VOTRIENT (pazopanib hydrochloride). It does not contain all of the available information.
It does not take the place of talking to your doctor or pharmacist.
You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on the medicine.
You can also download the most up to date leaflet from www.novartis.com.au.
The updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the expected benefits of you taking VOTRIENT against the risks this medicine could have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What VOTRIENT is used for
VOTRIENT contains the active substance pazopanib, which belongs to an anti-cancer group of medicines called protein kinase inhibitors.
VOTRIENT is used as a single agent to treat kidney cancer that is advanced or that has spread to other organs (metastatic). VOTRIENT is also used as a single agent to treat advanced Soft Tissue Sarcoma, which is a type of cancer that affects the supportive tissue of the body. It can occur in muscles, blood vessels, fat tissue or in other tissues that support, surround and protect organs.
VOTRIENT works by preventing the activity of proteins that are involved in the growth and spread of cancer cells.
Your doctor may have prescribed VOTRIENT for another reason.
This medicine is available only with a doctor's prescription.
VOTRIENT is not addictive.
Before you take VOTRIENT
When you must not take it
You must not take VOTRIENT if:
- You have ever had a severe allergic (hypersensitive) reaction to VOTRIENT (pazopanib hydrochloride).
Check with your doctor if you think this may apply to you. - You have ever had an allergic reaction to any of the ingredients listed toward the end of this leaflet.
(See "Ingredients") - The expiry date (EXP) printed on the pack has passed.
- The packaging is torn or shows signs of tampering.
Before you start to take it
Tell your doctor if you:
- Have heart disease
- Have had heart failure or a heart attack
- Have had prior blood clots in the vein or in a lung
- Have had prior collapse of a lung
- Have high blood pressure
- Have liver disease
- Have had problems with bleeding, blood clots or narrowing of the arteries
- If you have or have had an aneurysm (enlargement and weakening of a blood vessel wall) or a tear in a blood vessel wall.
- Have had stomach or bowel problems such as perforation (hole) or fistula (abnormal passages or tunnels leading out of the gut).
- Have thyroid problems
- Are going to have a surgical or dental procedure, or if you have had either recently.
Check with your doctor if you think any of these may apply to you.
Before you take VOTRIENT, your doctor will take blood samples to check for any liver problems. You may need extra tests to check that your heart and thyroid are working properly. Your doctor may decide to adjust your dose or stop treatment based on the results of these tests.
Do not breast-feed while taking VOTRIENT. It is not known whether the ingredients in VOTRIENT pass into breast milk and so may harm your baby.
Talk to your doctor about this.
Taking other medicines and VOTRIENT
Before you take VOTRIENT, tell your doctor, healthcare provider, or pharmacist if you are taking any other medicines, have taken any recently, or if you start new ones. This includes herbal medicines and other medicines you have bought without a prescription.
VOTRIENT can affect some other medicines, or they can affect VOTRIENT. Taking both together can make it more likely that you will have side effects. These medicines include:
- Clarithromycin, ketoconazole, itraconazole, rifampicin, telithromycin, voriconazole (used to treat fungal infections)
- Atazanavir, indinavir, nelfinavir, ritonavir, saquinavir (used to treat HIV)
- Nefazodone (used to treat depression)
- Simvastatin (used to treat high cholesterol levels)
- Medicines that reduce stomach acid (e.g. esomeprazole).
Tell your doctor, healthcare provider, or pharmacist if you take any of these medicines. Ask them if you are not sure whether your medicine is one of the medicines listed above. Some are not to be taken with VOTRIENT. For others, the dose or the time you take the medicine may need to be changed.
Taking VOTRIENT with food and drink
Take VOTRIENT on an empty stomach, either at least one hour before or at least two hours after food. VOTRIENT is affected by the food you eat. For details see "How do I take VOTRIENT?"
Do not drink grapefruit juice while you are being treated with VOTRIENT as this may increase the chance of side effects.
Children and adolescents
Votrient is not recommended for use in children and adolescents under 18 years.
Pregnancy and breast-feeding
VOTRIENT is not recommended if you are pregnant or breast-feeding.
If you are pregnant or breast-feeding, planning to get pregnant or think you might be pregnant, talk to your doctor about the risks and potential benefits of taking VOTRIENT. Your doctor will discuss with you the potential risks of taking VOTRIENT during pregnancy or breast-feeding.
Use a reliable method of contraception to avoid becoming pregnant while you are taking VOTRIENT and for 2 weeks after you stop treatment with it.
If you become pregnant or think you are pregnant, tell your doctor or healthcare provider immediately.
Men taking VOTRIENT
Male patients (including those who have had vasectomies) with female partners who are pregnant, possibly pregnant, or who could become pregnant should use condoms during sexual intercourse while taking VOTRIENT and for at least 2 weeks after the last dose.
Driving and using machines
VOTRIENT can have side effects such as fatigue, weakness and loss of energy that may affect your ability to drive.
Do not drive or use machines unless you are feeling well.
How do I take VOTRIENT?
Always take VOTRIENT exactly as your doctor, healthcare provider, or pharmacist has told you.
You should check with them if you are not sure.
How much VOTRIENT to take
The usual dose is 800 mg VOTRIENT, taken once a day. Your doctor may decide to give you two 400 mg tablets or four 200 mg tablets to make up the 800 mg dose.
How and when to take it
Swallow the tablets whole with water, one after the other, at about the same time each day.
Do not break or crush the tablets as this affects the way the medicine is absorbed and may increase the chance of side effects.
It is important that you take VOTRIENT on an empty stomach, either at least one hour before or at least two hours after food. Taking the drug with food increases the amount absorbed into the body, which may increase side effects.
Depending on your response to treatment, your doctor may recommend adjusting your dose or temporarily stopping your treatment.
If you forget to take VOTRIENT
Do not take a double dose to make up for a missed dose. Take the next dose at the scheduled time.
How long to take it for
Take VOTRIENT for as long as your doctor recommends.
Do not stop unless your doctor advises you to.
If you have any further questions on the use of this medicine, ask your doctor, pharmacist or healthcare provider.
If you take too much (Overdose)
Immediately telephone your doctor or Poisons Information Centre (call 131126) for advice if you think you or anyone else may have taken too much VOTRIENT, even if there are no signs of discomfort or poisoning. If possible, show them the VOTRIENT pack. If you are not sure what to do, contact your doctor or pharmacist.
While you are taking VOTRIENT
While you are taking VOTRIENT, your doctor will take blood samples to check for any liver or thyroid problems. Your doctor will also take blood and urine samples to check for any kidney problems. You will also have your blood pressure checked. Your doctor will periodically record your electrocardiogram (ECG) to check your heart's electrical conduction.
Your doctor will also check on any recent surgical or dental procedures to see if you are healing properly.
Things you must do
Tell your doctor if, for any reason, you have not taken your medicine exactly as directed. Otherwise, your doctor may think that it was not working as it should and change your treatment unnecessarily.
Things you must not do
Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
Do not use VOTRIENT to treat any other complaints unless your doctor says to.
Things to be careful of
Be careful driving, operating machinery or doing jobs that require you to be alert until you know how Votrient affects you.
Side effects
Like all medicines, VOTRIENT can cause side effects, although not everybody gets them.
Check with your doctor as soon as possible if you think you are experiencing any side effects or allergic reactions due to taking VOTRIENT, even if the problem is not listed below.
If they occur, they are most likely to be minor and temporary.
Serious side effects
Tell your doctor immediately if you experience any of the serious side effects listed below while taking VOTRIENT, as they may become life threatening or fatal.
- Signs of liver problems (abnormal liver function, liver failure), which may include: yellowing of your skin or the whites of your eyes (jaundice), dark urine, tiredness, nausea and/or vomiting, loss of appetite, pain on the right side of your stomach area (abdomen), bruising easily.
- Signs of sudden and severe rise in blood pressure (hypertensive crisis), which may include: severe chest pain, severe headache, blurred vision, confusion, nausea and/or vomiting, severe anxiety, shortness of breath, seizures, fainting.
- Signs of brain swelling (posterior reversible encephalopathy syndrome, reversible posterior leukoencephalopathy syndrome), which may include: loss of speech, blindness or changes in vision, seizures, confusion, headache, lack of energy, high blood pressure.
- Signs of lung inflammation (interstitial lung disease, pneumonitis) which may include: cough that will not go away, shortness of breath.
- Signs of heart problems such as abnormal heart rhythm (QT-prolongation, Torsade de Pointes), cardiac dysfunction/ heart failure, heart attack which may include: irregular or fast heartbeat, rapid fluttering of your heart, fainting, chest pain or pressure, pain in your arms, back, neck or jaw, shortness of breath, leg swelling.
- Signs of stroke, which may include: numbness or weakness on one side of your body, trouble talking, headache, dizziness.
- Signs of blood clots in your veins, especially in your legs (deep vein thrombosis), which may also travel to your lungs (pulmonary embolism) which may include: sharp chest pain, shortness of breath, rapid breathing, leg pain, swelling of your arms/hands or legs/feet.
- Signs of blood clots in the small blood vessels in the kidneys and brain accompanied by a decrease in red blood cells and cells involved in clotting (thrombotic microangiopathy) which may include: bruising easily, high blood pressure, fever, confusion, drowsiness, seizures, decrease in urine output.
- Signs of bleeding problems (haemorrhage) which may include: blood in your stool, black stool, blood in your urine, stomach pain, coughing and/or vomiting up blood.
- Signs of a tear in your stomach or intestinal wall (perforation) or the development of an abnormal connection between two parts of your digestive tract (fistula) which may include: severe stomach pain, nausea and/or vomiting, fever, bloody or foul-smelling drainage (pus) from an opening in your stomach area (abdomen) or near your anus.
- Signs of infections (which can become serious) may include: fever, flu-like symptoms such as cough, tiredness and body aches that do not go away, shortness of breath and/or wheezing, pain while urinating, cuts, scrapes or wounds that are red, warm, swollen or painful.
- Signs of Tumour Lysis Syndrome (TLS). This occurs when a large number of cancer cells die within a short period, releasing their contents in to the blood. When this happens levels of uric acid, potassium, and phosphorus rise faster than the kidneys can remove them. Symptoms may include nausea, vomiting, diarrhoea, muscle cramps or twitches, weakness, numbness or tingling, fatigue and not passing urine as frequently.
Possible side effects
- Possible side effects include the following listed below.
If these side effects become severe, please tell your doctor, pharmacist or healthcare provider.
Very common side effects
These may affect more than 1 in 10 people:
- Tumour pain
- Diarrhoea
- Feeling or being sick (nausea or vomiting)
- Loss of appetite
- Stomach pain or discomfort
- Indigestion
- High blood pressure
- Headache
- Loss of strength
- Lack of energy
- Weakness/loss of strength
- Changes in hair colour
- Weight loss
- Problems with taste
- Scaly red skin rash
- Skin reaction or pain on the palms of the hands or soles of the feet (including tingling, numbness, pain, swelling or reddening)
- Dizziness
- Cough
- Shortness of breath
- Chest pain
- Swelling of hands, ankles or feet
- Muscle pain
- Pain in the bones, muscles, ligaments, joints and tendons
- Mouth sores/ inflammation of the lining in the mouth
- Unusual hair loss or thinning
- Loss of skin pigmentation.
Very common side effect that may show up in your blood tests:
- Increase in some substances (enzymes) produced by the liver
- Protein in urine
- Under-active thyroid gland
- Decrease in albumin (a protein found in the blood).
Common side effects
These may affect up to 1 in 10 people:
- Difficulty sleeping
- Heart becomes less effective at pumping blood (cardiac dysfunction)
- Changes in the heart's electrical conduction (QT-prolongation)
- Heart attack
- Reduction of blood supply to the heart (angina)
- Severe bleeding in the lung
- Flatulence
- Nose bleed
- Dry skin
- Nail disorder
- Hoarseness
- Indigestion
- Blurred vision
- Chills
- Slow heart rate
- Sudden collapse of a lung, causing shortness of breath
- Severe bleeding (haemorrhage) in the lung
- Muscle spasms
- Chest pain, shortness of breath, leg pain, and swelling of the legs/feet. These could be signs of a blood clot in your body (thromboembolism). If the clot breaks off, it may travel to your lungs and this may be life threatening or even fatal
- Severe bleeding in digestive tract (stomach and intestine)
- Infections, with or without changes in white blood cells (cells that fight infection)
- Temporary reduction in blood supply to the brain (mini-stroke).
Common side effect that may show up in your blood or urine tests:
- Under-active thyroid gland
- Protein in urine
- Blood in the urine
- Abnormal liver function
- Decrease in the number of cells involved in blood clotting (thrombocytopenia)
- Low white blood cell count (neutropenia)
- Increase in bilirubin (a substance produced by the liver)
- Increase in gamma-glutamyl transpeptidase (a liver enzyme)
- Increase in albumin (a protein found in the blood)
- Increase in lipase (an enzyme from the pancreas).
Uncommon side effects
These may affect up to 1 in 100 people:
- Blood clots accompanied by a decrease in red blood cells and cells involved in clotting. These clots may harm organs such as the brain and kidneys.
- Abnormal connection between parts of the digestive tract (fistula)
- Stroke
- Severe bleeding in the brain
- Dangerous rapid fluttering of the heart (Torsade de Pointes)
- Hole (perforation) in digestive tract
- Abnormal connection between parts of the digestive tract (fistula)
- Sudden and severe rise in blood pressure which may be life-threatening
- Liver failure
- Sudden and severe rise in blood pressure which may be life-threatening (hypertensive crisis)
- Inflammation of the pancreas
- Separation or tear of the lining of the back part of the eye (retinal detachment or tear). This can result in blurry or impaired vision.
- skin wound with no healing tendency (skin ulcer)
Uncommon side effects that may show up in your blood tests
- Abnormal increase in the amount of haemoglobin in your blood.
Rare side effects
- Inflammation of the lung
- Swelling of the brain that may be associated with high blood pressure, headache, loss of speech or vision, and/or seizure, which may be life threatening.
- an enlargement and weakening of a blood vessel wall or a tear in a blood vessel wall (aneurysms and artery dissections)
Frequency unknown
- tumour lysis syndrome resulting from a fast breakdown of cancer cells
- liver failure
Tell your doctor immediately if you notice any of the following:
- Wheezing, swelling of the lips/mouth, difficulty in breathing, hayfever, lumpy rash (hives) or fainting. These could be a symptom of an allergic reaction.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
If you get side effects
Tell your doctor or pharmacist if any of the side effects listed become severe or troublesome, or if you notice any side effects not listed in this leaflet.
How do I store VOTRIENT?
Keep this medicine where children cannot see or reach it, such as in a locked cupboard.
Do not store above 30°C.
Do not leave in a car, on a window sill or in a bathroom.
Do not use VOTRIENT after the expiry date which is stated on the bottle.
Keep VOTRIENT tablets in the bottle until it is time to take them.
Disposal
If you have any unwanted tablets don't put them in wastewater or household rubbish. Ask your pharmacist how to dispose of tablets you do not need. This will help to protect the environment.
Product description
What VOTRIENT looks like
VOTRIENT are modified capsule-shaped, film-coated tablets, available in plastic bottles with a child resistant closure.
200 mg
Pink tablets with 'GS JT' debossed on one side. Available in packs of 30 tablets and 90 tablets.
400 mg
White tablets with 'GS UHL' debossed on one side. Available in packs of 30 tablets and 60 tablets.
Ingredients
VOTRIENT contains the active ingredient pazopanib hydrochloride. Each film-coated tablet contains the equivalent of either 200 mg or 400 mg pazopanib.
VOTRIENT also contains:
- Microcrystalline cellulose (E460)
- Povidone (E1201)
- Sodium starch glycolate
- Magnesium stearate (E572)
- Hypromellose (E464)
- Titanium dioxide (E171)
- Macrogol (E1521)
- Polysorbate 80
- Iron oxide red (E172) (200 mg tablets only).
Sponsor
VOTRIENT is supplied by:
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road,
Macquarie Park NSW 2113 Australia
Telephone 1 800 671 203
www.novartis.com.au
® Registered Trademark
© Copyright 2021
This leaflet was prepared in September 2021.
Australian Registration Numbers:
AUST R 161282: VOTRIENT 200 mg tablets
AUST R 161281: VOTRIENT 400 mg tablets.
Internal document code:
(vot170522c based on PI vot170522i)
Published by MIMS July 2022

 Table 3 presents the incidence of very common (> 10%) treatment-related adverse events in STS for patients receiving Votrient versus those on placebo.
Table 3 presents the incidence of very common (> 10%) treatment-related adverse events in STS for patients receiving Votrient versus those on placebo. Table 4 presents laboratory abnormalities occurring in ≥ 15% of patients who received Votrient in the pivotal RCC studies. Grades are based on the NCI CTCAE.
Table 4 presents laboratory abnormalities occurring in ≥ 15% of patients who received Votrient in the pivotal RCC studies. Grades are based on the NCI CTCAE.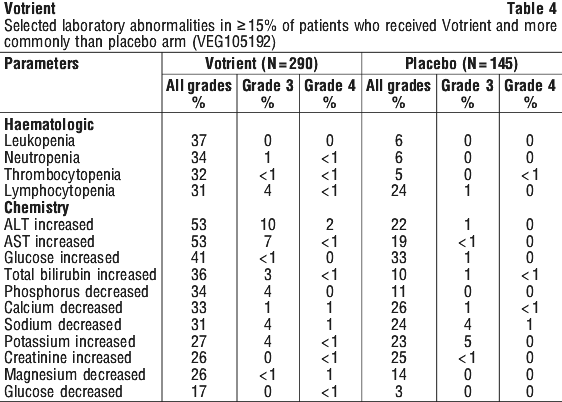




 For patients who responded to treatment, the median time to response was 11.9 weeks and the median duration of response was 58.7 weeks as per independent review.
For patients who responded to treatment, the median time to response was 11.9 weeks and the median duration of response was 58.7 weeks as per independent review. In VEG108844, health-related QoL was assessed using the following patient-reported questionnaires: Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), 19-item Functional Assessment of Cancer Therapy Kidney Symptom Index (FKSI-19), Cancer Therapy Satisfaction Questionnaire (CTSQ), and Supplementary Quality of Life Questionnaire (SQLQ). Overall compliance with SQLQ was 86%. Statistically significant differences in 11 of the 14 total domains favoured Votrient over sunitinib (p < 0.05), reflecting the adverse effects profile. The differences in fatigue, mouth/throat and hand/foot soreness and their limitations, feelings about sides effects and satisfaction with therapy were considered likely to be important (effect size > 0.20).
In VEG108844, health-related QoL was assessed using the following patient-reported questionnaires: Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F), 19-item Functional Assessment of Cancer Therapy Kidney Symptom Index (FKSI-19), Cancer Therapy Satisfaction Questionnaire (CTSQ), and Supplementary Quality of Life Questionnaire (SQLQ). Overall compliance with SQLQ was 86%. Statistically significant differences in 11 of the 14 total domains favoured Votrient over sunitinib (p < 0.05), reflecting the adverse effects profile. The differences in fatigue, mouth/throat and hand/foot soreness and their limitations, feelings about sides effects and satisfaction with therapy were considered likely to be important (effect size > 0.20). Similar to the assessments by independent radiology review, a clinically meaningful and statistically significant improvement in PFS based on investigator assessments was observed in the pazopanib arm compared with the placebo arm (HR: 0.39; 95% CI, 0.30 to 0.52, p < 0.001). See Figure 4.
Similar to the assessments by independent radiology review, a clinically meaningful and statistically significant improvement in PFS based on investigator assessments was observed in the pazopanib arm compared with the placebo arm (HR: 0.39; 95% CI, 0.30 to 0.52, p < 0.001). See Figure 4. The hazard ratio at the pre-specified interim analysis for overall survival in favour of pazopanib was not statistically significant; the median overall survival in the placebo arm was 10.4 months (95% CI 8.7 to 12.7) and was 11.9 months (95% CI 10.7 to 15.1) in the pazopanib arm; HR = 0.82 (97.87% CI: 0.59 to 1.14, p = 0.156). The overall survival in this study is potentially confounded due an imbalance of active treatments after disease progression, with more patients in the placebo arm receiving active therapy.
The hazard ratio at the pre-specified interim analysis for overall survival in favour of pazopanib was not statistically significant; the median overall survival in the placebo arm was 10.4 months (95% CI 8.7 to 12.7) and was 11.9 months (95% CI 10.7 to 15.1) in the pazopanib arm; HR = 0.82 (97.87% CI: 0.59 to 1.14, p = 0.156). The overall survival in this study is potentially confounded due an imbalance of active treatments after disease progression, with more patients in the placebo arm receiving active therapy. Molecular formula: C21H23N7O2S.HCl.
Molecular formula: C21H23N7O2S.HCl.