1 Name of Medicine
Diroximel fumarate.
2 Qualitative and Quantitative Composition
Vumerity capsules contain 231 mg of the active ingredient diroximel fumarate.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Enteric capsules.
Vumerity is supplied as enteric coated minitablets enclosed within white hypromellose capsules printed with "DRF 231 mg" in black ink on the capsule body.
4.1 Therapeutic Indications
Vumerity is indicated in patients with relapsing multiple sclerosis to reduce the frequency of relapses and to delay the progression of disability.
4.2 Dose and Method of Administration
The starting dose for Vumerity is 231 mg twice a day orally. After 7 days, the dose should be increased to the maintenance dose of 462 mg (administered as two 231 mg capsules) twice a day orally. Temporary dose reductions to 231 mg twice a day may be considered for individuals who do not tolerate the maintenance dose. Within 4 weeks, the recommended dose of 462 mg twice a day should be resumed. Discontinuation of Vumerity should be considered for patients unable to tolerate return to the maintenance dose.
Vumerity should be swallowed whole and intact. Vumerity should not be crushed or chewed and the capsule contents should not be sprinkled on food. Vumerity can be taken with or without food.
If a patient misses a dose, a double dose should not be taken. The patient may take the missed dose only if they leave 4 hours between doses. Otherwise, the patient should wait until the next scheduled dose.
Administration of 325 mg non-enteric coated aspirin prior to Vumerity dosing may reduce the occurrence and severity of flushing (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Renal or hepatic impairment.
A single-dose clinical study of Vumerity was conducted in patients with mild, moderate, and severe renal impairment. Degree of renal impairment had no effect on exposure to monomethyl fumarate (MMF). Therefore, no dosage adjustment is necessary in patients with renal impairment (see Section 5.2 Pharmacokinetic Properties). Long-term safety of Vumerity has not been studied in patients with moderate or severe renal impairment. The adverse event profile in subjects with mild renal impairment did not identify new safety concerns as compared to subjects with no renal impairment in EVOLVE-MS-1, the 96-week, open label study with Vumerity (see Section 5.2 Pharmacokinetic Properties).
Vumerity has not been studied in patients with hepatic impairment. Based on clinical pharmacology studies, no dose adjustments are needed.4.3 Contraindications
Vumerity is contraindicated in patients with known hypersensitivity to diroximel fumarate, any excipients in this product, or other fumaric acid derivatives.
Suspected or confirmed Progressive Multifocal Leukoencephalopathy (PML).
4.4 Special Warnings and Precautions for Use
Vumerity and Tecfidera are metabolised to monomethyl fumarate upon oral administration (see Section 4.2 Dose and Method of Administration). The risks associated with Vumerity are expected to be similar to those reported for dimethyl fumarate, even though not all the risks listed below have been observed specifically with Vumerity.
Infection.
Decreases in lymphocyte counts observed in patients treated with Tecfidera in clinical trials were not associated with increased frequencies of infections. However, due to the risk of serious, possibly fatal infection, patients who develop lymphopenia as a result of treatment with Vumerity require close monitoring. Patients should be instructed to report symptoms of infection to their physician. For patients with signs and symptoms of serious infections, interrupting treatment with Vumerity should be considered until the infection(s) resolves.
Serious cases of herpes zoster have occurred with Tecfidera, including disseminated herpes zoster, herpes zoster ophthalmicus, herpes zoster meningoencephalitis and herpes zoster meningomyelitis. These events may occur at any time during treatment. Monitor patients on Vumerity for signs and symptoms of herpes zoster. If herpes zoster occurs, appropriate treatment for herpes zoster should be administered. Consider withholding Vumerity treatment in patients with serious infections until the infection has resolved (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
Lymphopenia.
Vumerity may decrease lymphocyte counts (see Section 4.8 Adverse Effects (Undesirable Effects)). In EVOLVE-MS-1, the 96-week, open label clinical study with Vumerity, Vumerity was discontinued in patients with confirmed lymphocyte counts < 0.5 x 109/L which persisted for ≥ 4 weeks. In this study, 14.1% of patients (n=147) had lymphocyte counts ≥ 0.5 x 109/L and < 0.8 x 109/L for at least 6 months, 1.8% (n=19) of patients discontinued Vumerity due to confirmed lymphocyte counts < 0.5 x 109/L which persisted for ≥ 4 weeks, and an additional 0.6% (n=6) of patients who discontinued treatment with Vumerity due to low lymphocyte or leukocyte counts had at least one lymphocyte value of < 0.5 X 109/L.
In the Tecfidera multiple sclerosis (MS) placebo-controlled trials, mean lymphocyte counts decreased by approximately 30% during the first year of treatment with Tecfidera and then remained stable. WBC counts < 3.0 x 109/L and lymphocyte counts < 0.5 x 109/L were reported in 6 to 7% of subjects given Tecfidera. Prior to initiating treatment with Vumerity, a recent complete blood count (CBC) including lymphocytes (i.e. within 6 months) is recommended. A CBC, including lymphocytes, is also recommended, after 6 months of treatment and every 6 to 12 months thereafter, and as clinically indicated. In clinical studies of Tecfidera, 9% of patients had lymphocyte counts ≥ 0.5 x 109/L and < 0.8 x 109/L for at least six months. 2% experienced lymphocyte counts < 0.5 x 109/L, for at least six months and in this group the majority of lymphocyte counts remained < 0.5 x 109/L with continued therapy. In controlled and uncontrolled clinical studies, the mean time for lymphocyte counts to return to normal after discontinuing Tecfidera treatment was 4.7 weeks in patients without prolonged, severe lymphopenia and 29 weeks in patients with prolonged, severe lymphopenia (patients with lymphocyte counts < 0.5 x 109/L for six months or greater).
Consider interruption of Vumerity in patients with lymphocyte counts < 0.5 x 109/L persisting for more than six months. Lymphocytes counts should be followed until recovery. Assess the benefit/risk in patients who experience moderate lymphopenia for more than 6 months.
Interrupting treatment should be considered in patients with serious infections until the infection(s) resolved. Vumerity has not been studied in patients with pre-existing low lymphocyte counts and caution should be exercised when treating these patients.
Magnetic resonance imaging (MRI).
Before initiating treatment, a baseline MRI should be available (usually within 3 months) as a reference. The need for further MRI scanning should be considered in accordance with national recommendations. MRI imaging may be considered as part of increased vigilance in patients considered at increased risk of PML. In case of clinical suspicion of PML, MRI should be performed immediately for diagnostic purposes.
Progressive multifocal leukoencephalopathy.
PML has been reported in patients treated with dimethyl fumarate. PML is an opportunistic infection caused by John Cunningham virus (JCV), which may be fatal or result in severe disability.
PML cases have occurred with dimethyl fumarate and other medicinal products containing fumarates in the setting of lymphopenia (lymphocyte counts below lower limit of normal [LLN]). Prolonged moderate to severe lymphopenia appears to increase the risk of PML with dimethyl fumarate, however, risk cannot be excluded in patients with mild lymphopenia.
Additional factors that might contribute to an increased risk for PML in the setting of lymphopenia are:
duration of Vumerity therapy. Cases of PML have occurred after approximately 1 to 5 years of dimethyl fumarate treatment, although the exact relationship with duration of treatment is unknown;
profound decreases in CD4+ and especially in CD8+ T cell counts, which are important for immunological defense (see Section 4.8 Adverse Effects (Undesirable Effects)); and
prior immunosuppressive or immunomodulatory therapy (see below).
Physicians should evaluate their patients to determine if the symptoms are indicative of neurological dysfunction and, if so, whether these symptoms are typical of MS or possibly suggestive of PML.
At the first sign or symptom suggestive of PML, Vumerity should be withheld and appropriate diagnostic evaluations, including determination of JCV DNA in cerebrospinal fluid (CSF) by quantitative polymerase chain reaction (PCR) methodology, need to be performed. The symptoms of PML may be similar to an MS relapse. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. Physicians should be particularly alert to symptoms suggestive of PML that the patient may not notice. Patients should also be advised to inform their partner or caregivers about their treatment, since they may notice symptoms that the patient is not aware of.
PML can only occur in the presence of a JCV infection. It should be considered that the influence of lymphopenia on the accuracy of serum anti-JCV antibody testing has not been studied in dimethyl fumarate or Vumerity treated patients. It should also be noted that a negative anti-JCV antibody test (in the presence of normal lymphocyte counts) does not preclude the possibility of subsequent JCV infection.
If a patient develops PML, Vumerity must be permanently discontinued.
Prior treatment with immunosuppressive or immunomodulating therapies.
No studies have been performed evaluating the efficacy and safety of diroximel fumarate when switching patients from other disease modifying therapies. The contribution of prior immunosuppressive therapy to the development of PML is possible.
PML cases have occurred in patients who had previously been treated with natalizumab, for which PML is an established risk. Physicians should be aware that cases of PML occurring following recent discontinuation of natalizumab may not have lymphopenia.
In addition, a majority of confirmed PML cases with dimethyl fumarate occurred in patients with prior immunomodulatory treatment.
When switching patients from another disease modifying therapy to Vumerity, the half-life and mechanism of action of the other therapy should be considered in order to avoid an additive immune effect while at the same time, reducing the risk of reactivation of MS. A complete blood count is recommended prior to treatment initiation and regularly during treatment (see Lymphopenia above).
Flushing.
In dimethyl fumarate pivotal clinical trials, 3 patients out of a total of 2,560 patients treated with dimethyl fumarate experienced serious flushing symptoms that were probable hypersensitivity or anaphylactoid reactions. These adverse reactions were not life-threatening but led to hospitalisation. Prescribers and patients should be alert to this possibility in the event of severe flushing reactions with Vumerity (see Section 4.2 Dose and Method of Administration; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 4.8 Adverse Effects (Undesirable Effects)).
Data from healthy volunteer studies suggest that dimethyl fumarate-associated flushing is likely to be prostaglandin mediated. A short course of treatment with 75 mg non-enteric coated aspirin may be beneficial in patients affected by intolerable flushing (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). In two healthy volunteer studies, the occurrence and severity of flushing over the dosing period was reduced.
Anaphylactic reactions.
Cases of anaphylaxis have been reported following Tecfidera administration. These reactions generally occurred after the first dose, but may occur at any time during treatment, and may be serious and life threatening. Patients should be instructed to discontinue Vumerity and seek immediate medical care if they experience signs or symptoms of anaphylaxis. Treatment should not be restarted (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
Vaccination.
Patients taking Vumerity may receive non-live vaccines (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). The safety of administration of live attenuated vaccines during treatment with Vumerity has not been evaluated in clinical trials. Live vaccines have a potential risk of clinical infection and are not recommended during treatment with Vumerity.
Serious gastrointestinal reactions.
Serious gastrointestinal reactions, including perforation, ulceration, haemorrhage, and obstruction, some with fatal outcomes, have been reported in the post-marketing setting with the use of fumaric acid esters, including Vumerity, with or without concomitant aspirin use. The majority of these events have occurred within 6 months of fumaric acid ester treatment initiation. In controlled clinical trials, the incidence of serious gastrointestinal adverse events was 1% in patients treated with Vumerity; these events, none of which were fatal, included vomiting (0.3%) and abdominal pain (0.3%) (see Section 4.8 Adverse Effects (Undesirable Effects)).
Effects on renal function.
In clinical trials with patients with MS, adverse events of proteinuria (proteinuria, microalbuminuria and urine albumin present) were reported at slightly higher frequencies in patients treated with Tecfidera compared to patients that received placebo. The significance of these clinical observations is not known at this time.
Prior to initiating treatment with Vumerity, urinalysis should be available (within 6 months prior to starting therapy). During treatment, urinalysis is recommended annually and as clinically indicated.
The use of Vumerity in patients who receive chronic treatment with medications that are associated with potential nephrotoxic risk (e.g. aminoglycosides, diuretics, NSAIDs, lithium) has not been evaluated. Therefore, caution should be exercised if Vumerity is used in patients receiving chronic treatment with such medications.
Effects on liver function.
In clinical studies, high post-baseline values were observed mostly for ALT and AST, and more frequently in the Vumerity group (25.9% and 15.8%) vs. the Tecfidera group (16.4% and 9.0%) respectively.
Drug-induced liver injury, including liver enzyme increase (≥ 3 x upper limit of normal (ULN)) and elevation of total bilirubin levels (≥ 2 x ULN) can result from treatment with dimethyl fumarate. The time to onset can be directly, several weeks or longer. Resolution of the adverse reactions has been observed after treatment was discontinued. Assessment of serum aminotransferases (e.g. alanine aminotransferase (ALT), aspartate aminotransferase (AST)) and total bilirubin levels are recommended prior to treatment initiation and during treatment as clinically indicated.
Use in hepatic impairment.
Based on pharmacokinetic findings, no dose adjustment is necessary for patients with hepatic impairment.
Diroximel fumarate has not been studied in patients with hepatic impairment. Caution should be used when considering treatment in patients with severe hepatic impairment (see Section 4.2 Dose and Method of Administration, Renal or hepatic impairment; Section 5.2 Pharmacokinetic Properties, Hepatic impairment).
Treatment initiation.
Treatment should be started gradually to reduce the occurrence of flushing and gastrointestinal adverse reactions (see Section 4.2 Dose and Method of Administration).
Fanconi syndrome.
Cases of Fanconi syndrome have been reported for a medicinal product containing dimethyl fumarate in combination with other fumaric acid esters. Early diagnosis of Fanconi syndrome and discontinuation of Vumerity treatment are important to prevent the onset of renal impairment and osteomalacia, as the syndrome is usually reversible. The most important signs are: proteinuria, glucosuria (with normal blood sugar levels), hyperaminoaciduria and phosphaturia (possibly concurrent with hypophosphatemia). Progression might involve symptoms such as polyuria, polydipsia and proximal muscle weakness. In rare cases, hypophosphataemic osteomalacia with non-localised bone pain, elevated alkaline phosphatase in serum and stress fractures may occur. Importantly, Fanconi syndrome can occur without elevated creatinine levels or low glomerular filtration rate. In case of unclear symptoms, Fanconi syndrome should be considered and appropriate examinations should be performed.
Use in the elderly.
There are limited data available for the use of Vumerity in patients aged 65 years and over, therefore it is unknown whether elderly patients respond differently to younger patients.
Paediatric use.
The safety and effectiveness of Vumerity in paediatric patients with MS below the age of 18 have not been established. In a juvenile rat study, effects on bone geometry and density were seen in animals treated orally with 600 mg/kg/day diroximel fumarate. Exposures to monomethyl fumarate and HES were 6 times the AUC at the MRHD of diroximel fumarate.
Effects on laboratory tests.
There are no data available on whether Vumerity interferes with laboratory tests.4.5 Interactions with Other Medicines and Other Forms of Interactions
During treatment with Vumerity, simultaneous use of other fumaric acid derivatives (topical or systemic) should be avoided as such clinical scenarios have not been studied. Do not administer Vumerity concomitantly with dimethyl fumarate.
In humans, diroximel fumarate is extensively metabolised by esterases before it reaches the systemic circulation and further metabolism occurs through the tricarboxylic acid (TCA) cycle, with no involvement of the cytochrome P450 (CYP) system. Potential drug interaction risks were not identified from in vitro CYP-inhibition and induction studies, or studies of the protein binding of diroximel fumarate and its active metabolite monomethyl fumarate.
A pharmacokinetic (PK) study with digoxin has been performed with diroximel fumarate (Vumerity) and showed that diroximel fumarate and its metabolites did not inhibit P-glycoprotein (P-gp) in vivo. The major metabolites of diroximel fumarate, the active monomethyl fumarate and the inactive 2-hydroxyethyl succinimide HES, did not inhibit P-gp, BCRP, MATE1, MATE2-K, OAT1, OAT3, OCT1, OCT2, OATP1B1 or OATP1B3 in vitro.
Based on in vitro data, MMF is not predicted to inhibit BSEP and HES is not expected to inhibit OCT1 at the maximum clinical dose. HES was not a substrate for P-gp, BCRP, MATE1, MATE2K, OAT1, OAT3 or OCT2 and MMF is not a substrate for P-gp.
Although not studied with Vumerity, a PK study with a combined oral contraceptive has been performed with dimethyl fumarate. There were no relevant effects of dimethyl fumarate on the PK profile of norelgestromin and ethinyl oestradiol. No interaction studies have been performed with oral contraceptives containing other progestogens; however, an effect of Vumerity on their exposure is not expected.
Commonly used drugs in patients with MS, intramuscular (IM) interferon beta-1a and glatiramer acetate (GA), were clinically tested for potential drug-interactions with Tecfidera and did not alter the pharmacokinetic profile of Tecfidera. Aspirin (non-enteric coated), 325 mg, when administered approximately 30 minutes before Tecfidera, did not alter the pharmacokinetic profile of Tecfidera.
Patients treated with Tecfidera were able to mount an effective immune response to inactivated neoantigen (first vaccination), recall antigen (re-exposure), or polysaccharide antigen in a clinical study in patients with relapsing forms of MS. This response was comparable to patients treated with non-pegylated interferons. Patients taking Vumerity may receive non-live vaccines. No clinical data are available on the efficacy and safety of live attenuated vaccines in patients taking Vumerity.
Diroximel fumarate has not been studied in combination with anti-neoplastic or immunosuppressive therapies and caution should, therefore, be used during concomitant administration. In MS clinical studies, the concomitant treatment of relapses with a short course of intravenous corticosteroids was not associated with a clinically relevant increase of infection.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Data from nonclinical studies do not suggest that Vumerity would be associated with an increased risk of reduced fertility.
Diroximel fumarate did not impair fertility in rats when administered orally to males up to 400 mg/kg/day and to females up to 450 mg/kg/day (monomethyl fumarate and HES exposures were each up to approximately 6x in males and 7x in females, relative to exposure at the maximum recommended human dose [MRHD] of diroximel fumarate on an AUC basis).
(Category B3)
There are no adequate and well-controlled studies in pregnant women. Vumerity is not recommended during pregnancy and in women of childbearing potential not using appropriate contraception. The effects of Vumerity on labour and delivery are unknown.
Animal developmental toxicity studies indicated adverse embryofetal effects, likely secondary to maternal toxicity.
Oral administration of diroximel fumarate (up to 400 mg/kg/day) to pregnant rats throughout organogenesis resulted in lower fetal body weights and an increase in fetal skeletal variations at the highest dose tested, which was associated with maternal toxicity. Plasma exposures (AUC) for MMF and HES (the major circulating drug-related compound in humans) at the no effect dose (100 mg/kg/day) for adverse effects on embryofetal development were each approximately 2 times those in humans at the MRHD.
Oral administration of diroximel fumarate (up to 350 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in an increase in fetal skeletal malformations at the mid and high doses and lower fetal body weight and increases in embryofetal death and fetal skeletal variations at the highest dose tested. The mid and high dose was associated with maternal toxicity. Plasma exposures (AUC) for MMF and HES at the no-effect dose (50 mg/kg/day) for adverse effects on embryofetal development were 1.4x and 0.7x, respectively, those in humans at the MRHD.
Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed and if the potential benefit justifies the potential risk to the fetus.
It is not known whether this drug is excreted in human milk. HES was detected in the circulation of breast-fed rat pups following maternal exposure to diroximel fumarate. A risk to newborns/infants cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue treatment with Vumerity, taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman. Caution should be exercised when Vumerity is administered to a nursing woman.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been conducted.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
Upon oral administration, Vumerity and Tecfidera are rapidly metabolised to monomethyl fumarate before they reach the systemic circulation. Adverse reactions are expected to be similar between Vumerity and Tecfidera once metabolised.
There are 2 Phase 3 clinical trials for Vumerity in patients with relapsing-remitting MS (RRMS): EVOLVE-MS-1, an open-label, 2-year safety study; and EVOLVE MS 2, randomised, double-blind study comparing the GI tolerability of Vumerity to Tecfidera. In these studies, the adverse reaction profile observed with Vumerity was similar to what was seen with the Tecfidera clinical trial experience.
The most common adverse reactions (incidence ≥ 10% and > 2% than placebo) for Tecfidera were flushing and gastrointestinal (GI) events (i.e. diarrhoea, nausea, abdominal pain, upper abdominal pain).
The most commonly reported adverse events leading to discontinuation (incidence > 1%) in patients treated with Tecfidera were flushing (3%) and gastrointestinal events (4%). It has been demonstrated that Vumerity has fewer and less severe GI adverse events than dimethyl fumarate (see Table 3). Based on patient reported outcomes, Vumerity demonstrated less severe GI events and fewer days of self-reported GI symptoms compared with Tecfidera (see EVOLVE-MS-2 study description below).
In placebo-controlled and uncontrolled clinical studies, a total of 2513 patients have received Tecfidera and been followed for periods up to 12 years with an overall exposure equivalent to 11,318 person-years. A total of 1169 patients have received at least 5 years of treatment with Tecfidera, and 426 patients have received at least 10 years of treatment with Tecfidera.
In the two Phase 3 placebo-controlled trials, 1529 patients received Tecfidera with an overall exposure of 2371 person-years (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The adverse reactions presented in the tables below are based on safety information from 769 patients treated with Tecfidera 240 mg twice a day and 771 patients treated with placebo. Based on the frequency, Table 1 presents common and very common adverse reactions and Table 2 presents the actual frequency in the Tecfidera arm as compared to the placebo arm.
The adverse reactions are presented as MedDRA preferred terms under the MedDRA system organ class.
The incidence of the adverse reactions below is expressed according to the following categories: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1, 000 to < 1/100); rare (≥ 1/10, 000 to < 1/1,000); very rare (< 1/10,000).

 Other relevant ADRs (< 2% difference) include: gastroenteritis, gastritis, gastrointestinal disorder, burning sensation, feeling hot, alanine aminotransferase increased, proteinuria, white blood cell count decreased and leucopenia.
Other relevant ADRs (< 2% difference) include: gastroenteritis, gastritis, gastrointestinal disorder, burning sensation, feeling hot, alanine aminotransferase increased, proteinuria, white blood cell count decreased and leucopenia.
In EVOLVE-MS-1, the Phase 3, open-label study of Vumerity, 1057 subjects received at least 1 dose of Vumerity, and 912 subjects had > 12 months of exposure. Data from this study showed drug-related adverse reactions were similar to those seen in Tecfidera. The most common adverse reactions from this study (≥ 10%) were flushing (27.2%), lymphopenia (11.7%) and diarrhoea (10.3%). Additionally, HES, an inactive metabolite of Vumerity that is renally excreted did not appear to have any clinically relevant impact on the safety profile of Vumerity.
In EVOLVE-MS-2, a total of 506 patients were randomised; 504 were dosed with at least 1 dose of study drug and included in the safety population: Vumerity (N=253) or Tecfidera (N=251). Vumerity met the primary endpoint demonstrating a significant reduction in the number of days with an Individual Gastrointestinal Symptom and Impact Scale (IGISIS, a patient self-assessment tool for GI symptom severity and impact) score of ≥ 2 relative to exposure (adjusted rate ratio [95% confidence interval, CI]) of 0.54 ([0.39 - 0.75], p=0.0003), representing a 46% reduction with Vumerity compared to Tecfidera. In this study, overall adverse events were 34.8% and 49%, respectively. Treatment discontinuations were 1.6% and 6%, respectively, and the differences in these numbers were driven by discontinuations for GI tolerability reasons (0.8% and 4.8%, respectively). Treatment-related adverse events of ≥ 5% for Vumerity or Tecfidera, respectively, that demonstrated a numerical difference between the 2 groups of > 2% were flushing (32.8% versus 40.2%), diarrhoea (13.8% versus 18.7%), nausea (13.4% versus 17.9%), upper abdominal pain (6.3% versus 13.9%), abdominal pain (5.5% versus 9.6%), vomiting (3.2% versus 7.6%) (see Table 3).

Paediatric patients.
There are no paediatric data available for Vumerity.
Description of selected adverse events.
The adverse reaction profile of Vumerity is expected to be similar to Tecfidera.
Flushing.
Flushing with Vumerity use is expected to be similar to Tecfidera. In EVOLVE-MS-1, < 1% of patients treated with Vumerity discontinued due to flushing. The incidence of serious flushing, which may be characterised by generalised erythema, rash, and/or pruritus was seen in < 1% of patients treated with Vumerity in EVOLVE-MS-1 (see Section 4.2 Dose and Method of Administration). In EVOLVE-MS-2, flushing was reported in 32.8% of Vumerity patients and 40.2% of Tecfidera patients, and there were no discontinuations due to flushing.
In Tecfidera, the incidence of patients with flushing events (e.g. warmth, redness, itching, burning sensation) was higher early in the course of treatment (primarily in month 1) and decreased over time, which might indicate that this symptom became less prevalent with continued use. In patients with flushing, the majority had flushing events that were mild or moderate in severity.
Gastrointestinal.
GI tolerability in patients treated with Vumerity was directly compared to Tecfidera in EVOLVE-MS-2. In this study, 0.8% (n=2) treated with Vumerity discontinued treatment due to GI events, as compared with 4.8% (n=12) for Tecfidera. There were no serious GI events for either Vumerity or Tecfidera in this study. In EVOLVE-MS-1, < 1% of patients treated with Vumerity discontinued due to GI events, and < 1% of patients had serious GI events.
The incidence of patients with GI events (e.g. nausea, vomiting, diarrhoea, abdominal pain, upper abdominal pain and dyspepsia) was higher early in the course of treatment (primarily in month 1) and decreased over time in patients treated with Tecfidera compared with placebo.
Hepatic transaminases.
In EVOLVE-MS-2, the majority of patients with elevations had hepatic transaminases that were < 3 times ULN and did not require dose adjustment or termination. Elevations in hepatic aminotransferase ≥ 3 times ULN and ≥ 5 times ULN, respectively, were seen in 0.8% (n=2) and 0.4% (n=1) of patients for Vumerity and 1.6% (n=4) and 0.4% (n=1) of patients for Tecfidera. Treatment interruptions due to elevated hepatic transaminases were seen in 0.8% (n=2) of patients treated with Vumerity and 0.4% (n=1) of patients treated with Tecfidera. In EVOLVE-MS-1, < 1% of patients discontinued treatment due to elevations in hepatic transaminases. Elevations in transaminases ≥ 3 times ULN with concomitant elevations in total bilirubin > 2 times ULN have not been observed with Vumerity.
Subjects discontinued treatment if ALT or AST values remained > 3x ULN for ≥ 4 weeks in the 2-year study EVOLVE-MS-1 and ≥ 2 weeks in the 5-week study EVOLVE-MS-2. In both studies, transient increases in liver transaminases were observed with treatment initiation, with greater mean increases from baseline in ALT than in AST, which subsided with continued treatment. They were not associated with symptoms of liver injury or disease. As DRF metabolism does not involve CYP enzymes, it is considered that is unlikely to cause clinically relevant PK interactions with drugs metabolised by CYP enzymes.
Elevations in transaminases ≥ 3 times ULN with concomitant elevations in total bilirubin > 2 times ULN were not observed during placebo-controlled studies with Tecfidera but have been observed in the post-marketing experience (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience below).
Haematological.
The impact of Vumerity use on lymphocytes is expected to be similar to Tecfidera. In the placebo-controlled studies for Tecfidera, most patients (> 98%) had normal lymphocyte values prior to initiating treatment. Upon treatment with Tecfidera, lymphocyte counts decreased over the first year with a subsequent plateau. On average, lymphocyte counts decreased by approximately 30% of baseline value. Mean and median lymphocyte counts remained within normal limits. Patients with lymphocyte counts < 0.5 x 109/L were observed in < 1% of patients treated with placebo and 6% of patients treated with Tecfidera. In controlled and uncontrolled clinical studies, 2% of patients experienced lymphocyte counts < 0.5 x 109/L for at least six months. In these patients, the majority of lymphocyte counts remained < 0.5 x 109/L with continued therapy.
The incidence of infections (58% vs 60%) and serious infections (2% vs 2%) was similar in patients treated with placebo or Tecfidera, respectively. An increased incidence of infections and serious infections was not observed in patients with lymphocyte counts < 0.8 x 109/L or 0.5 x 109/L. A transient increase in mean eosinophil counts was seen during the first 2 months of therapy.
In EVOLVE-MS-1, 1.8% (n=19) of patients discontinued Vumerity due to confirmed lymphocyte counts < 0.5 x 109/L which persisted for ≥ 4 weeks, and an additional 0.6% (n=6) of patients who discontinued treatment with Vumerity due to low lymphocyte or leukocyte counts had at least one lymphocyte value of < 0.5 x 109/L. There were no discontinuations for infections related to Vumerity treatment.
Post-marketing experience.
In post marketing experience for Tecfidera, hypersensitivity reactions including urticaria, angioedema, and difficulty breathing have been reported following Tecfidera administration. Cases of anaphylaxis have also been reported (see Section 4.4 Special Warnings and Precautions for Use, Anaphylactic reactions).
Progressive multifocal leukoencephalopathy has occurred in the setting of lymphopenia (< 0.91 x 109/L) following Tecfidera administration. These PML cases have occurred predominantly in the setting of prolonged moderate to severe lymphopenia. (See Section 4.4 Special Warnings and Precautions for Use, Progressive multifocal leukoencephalopathy).
Liver function abnormalities (elevations in transaminases ≥ 3 times ULN with concomitant elevations in total bilirubin > 2 times ULN) have been reported following Tecfidera administration in post marketing experience. These abnormalities resolved upon treatment discontinuation over a varying period of time. Therefore, ongoing monitoring of LFTs is recommended in patients being treated with Vumerity, as clinically indicated.
Herpes zoster infection has been reported with Tecfidera administration in post marketing experience. The majority of cases were non-serious (see Section 4.4 Special Warnings and Precautions for Use, Infection).
Rhinorrhea and alopecia have been reported with Tecfidera administration in post marketing experience.4.9 Overdose
Cases of overdose with Tecfidera have been reported. There are no known therapeutic interventions to enhance elimination of Vumerity nor is there a known antidote. In the event of overdose, it is recommended that symptomatic supportive treatment be initiated as clinically indicated.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
The pathophysiology of MS is multifaceted and propagated through ongoing inflammatory and neurodegenerative stimuli, mediated at least in part by toxic oxidative stress. Preclinical studies indicate that diroximel fumarate pharmacodynamic responses appear to be mediated, at least in part, through activation of the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcriptional pathway, which is the primary cellular defense system for responding to a variety of potentially toxic stimuli through up-regulation of antioxidant response genes. Dimethyl fumarate reduces inflammatory responses in both peripheral and central cells and promotes cytoprotection of central nervous system cells against toxic oxidative damage, demonstrating effects on pathways known to exacerbate MS pathology. Dimethyl fumarate has also been shown to up-regulate Nrf2-dependent antioxidant genes in patients, confirming clinical pharmacodynamic activity in humans. Vumerity and dimethyl fumarate undergo rapid hydrolysis prior to systemic circulation by esterases and are converted to the primary active metabolite, MMF. However, the mechanism by which Vumerity and dimethyl fumarate exert therapeutic effects in MS is not fully understood.
Pharmacodynamic effects.
Activation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway.
The diroximel fumarate and dimethyl fumarate mechanism of action appears to be mediated, at least in part, through activation of the Nrf2 anti-oxidant response pathway. Biological response markers of Nrf2 activation (e.g. NAD(P)H dehydrogenase, quinone 1 [NQO1]) are detected at elevated levels in blood from patients with MS following 12 or 48 weeks of oral dosing with dimethyl fumarate. These clinical data appear to be consistent with preclinical studies demonstrating dimethyl fumarate-dependent up-regulation of Nrf2 antioxidant response genes in multiple tissue types. The relationships between blood NQO1 levels and the mechanism(s) by which dimethyl fumarate exerts its effects in MS are unknown.
Effects on immune system.
In preclinical and clinical studies, dimethyl fumarate demonstrates anti-inflammatory and immunomodulatory properties. Dimethyl fumarate and monomethyl fumarate (the active metabolite of diroximel fumarate and dimethyl fumarate) significantly reduce immune cell activation and subsequent release of pro-inflammatory cytokines in response to inflammatory stimuli, and moreover affects lymphocyte phenotypes through a down-regulation of proinflammatory cytokine profiles (TH1, TH17), and biases towards anti-inflammatory production (TH2). Dimethyl fumarate demonstrates therapeutic activity in multiple models of inflammatory and neuroinflammatory injury, and also appears to promote improvement in blood brain barrier integrity. All of these anti-inflammatory effects appear consistent with the significant clinical activity of diroximel fumarate and dimethyl fumarate in reducing brain lesions and relapses in MS patients.
Effects on central nervous system.
In preclinical and clinical studies monomethyl fumarate is able to penetrate into the central nervous system where it promotes cyto- and neuro-protective responses. Dimethyl fumarate and monomethyl fumarate significantly improve cell viability after oxidative challenge in primary cultures of astrocytes and neurons, suggesting monomethyl fumarate and dimethyl fumarate directly prevent neurodegeneration in response to toxic stress. Acute neurotoxic injury models and genetic models of neurodegenerative disease confirm that dimethyl fumarate provides therapeutic benefit in reducing neuronal and functional damage resulting from various types of toxic stimuli and other forms of cellular stress inherent in neurodegenerative disease states. These preclinical data combined with imaging and functional endpoints from clinical studies suggest dimethyl fumarate and diroximel fumarate may promote a neuroprotective benefit in the central nervous system.
Effects on gastrointestinal system.
In a double-blind clinical study comparing GI tolerability of diroximel fumarate versus dimethyl fumarate, diroximel fumarate demonstrated reduced incidence and severity of GI adverse events, as well as GI adverse events leading to treatment discontinuation, compared to dimethyl fumarate (see Section 4.8 Adverse Effects (Undesirable Effects)). The mechanism by which diroximel fumarate and dimethyl fumarate induce GI tolerability adverse events is unknown. Diroximel fumarate has a distinct chemical structure, and together with the data from clinical studies demonstrating an improvement in GI tolerability compared to dimethyl fumarate suggest the chemical structure of diroximel fumarate may contribute to a favourable GI tolerability profile.
Effect on cardiovascular system.
In a double-blind, placebo- and active-controlled thorough QT study in healthy volunteers, diroximel fumarate up to 2 times the recommended doses (924 mg twice a day) did not have a clinically relevant effect on QTc interval.
Effects of metabolite HES.
HES is a major inactive metabolite of diroximel fumarate. In in vitro studies, HES demonstrated no biological activities at concentrations similar to and exceeding those seen clinically and was not shown to interfere with biological activity of monomethyl fumarate. To assess the potential impact of HES on efficacy in vivo, diroximel fumarate was tested compared to dimethyl fumarate in a standard rat model of MS and diroximel fumarate and dimethyl fumarate were found to have similar efficacy, demonstrating that HES does not interfere with efficacy in vivo. In the 96-week clinical study of diroximel fumarate in patients with MS (EVOLVE-MS-1), HES did not appear to have any clinically relevant impact on the safety profile of diroximel fumarate.
Clinical trials.
Vumerity and Tecfidera are rapidly metabolised by esterases before they reach the systemic circulation to the same active metabolite, monomethyl fumarate, upon oral administration. The PK comparability of Vumerity to Tecfidera through the analysis of monomethyl fumarate exposure has been demonstrated (see Section 5.2 Pharmacokinetic Properties).
The clinical studies described in the following sections were conducted using Tecfidera.
The efficacy and safety of Tecfidera was demonstrated in three studies that evaluated Tecfidera taken either twice or three times a day in patients with relapsing-remitting multiple sclerosis (RRMS). The starting dose for Tecfidera was 120 mg twice or three times a day for the first 7 days, followed by an increase to either 240 mg twice or three times a day. Two of the three studies (Study 1 and Study 2) included patients with Expanded Disability Status Scale (EDSS) scores ranging from 0 to 5, who had experienced at least 1 relapse during the year prior to randomisation, or, within 6 weeks of randomisation had a brain Magnetic Resonance Imaging (MRI) demonstrating at least one gadolinium-enhancing (Gd+) lesion.
Study 1 (DEFINE) was a 2-year randomised, double-blind, placebo-controlled study in 1234 patients with RRMS who had not received interferon-beta or glatiramer acetate (GA) for at least the previous 3 months or natalizumab for at least the previous 6 months. Neurological evaluations were performed at baseline, every 3 months and at time of suspected relapse. MRI evaluations were performed at baseline, month 6, and year 1 and 2.
The primary endpoint in Study 1 was the reduction in the proportion of patients relapsed at 2 years. Patients were randomised to receive Tecfidera 240 mg twice a day (n=410), Tecfidera 240 mg three times a day (n=416), or placebo (n=408) for up to 2 years. Median age: 39 years, median years since diagnosis: 4.0 years and median EDSS score at baseline: 2.0. Median time on study was 84 weeks on 240 mg twice a day, 83 weeks on 240 mg three times a day and 85 weeks on placebo.
The proportion of patients relapsed was significantly lower in the group treated with Tecfidera than in the group treated with placebo at 2 years. Secondary endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of Gd-enhancing lesions, annualised relapse rate (ARR), and time to confirmed disability progression. Confirmed disability progression was defined as at least a 1 point increase from baseline EDSS (1.5 point increase for patients with baseline EDSS of 0) sustained for 12 weeks. Tecfidera had a clinically meaningful and statistically significant effect on all primary and secondary study endpoints. The 240 mg three times daily dose resulted in no additional benefit over the Tecfidera 240 mg twice daily dose. The results for this study are shown in Table 4.
 Study 2 (CONFIRM) was a 2-year multicentre, randomised, double-blind, placebo-controlled study which contained a rater-blinded (i.e. study physician/investigator assessing the response to study treatment is blinded) reference comparator of glatiramer acetate (GA) in 1417 patients with RRMS.
Study 2 (CONFIRM) was a 2-year multicentre, randomised, double-blind, placebo-controlled study which contained a rater-blinded (i.e. study physician/investigator assessing the response to study treatment is blinded) reference comparator of glatiramer acetate (GA) in 1417 patients with RRMS.
Patients had not received interferon-beta for at least the previous 3 months, natalizumab for at least the previous 6 months and had not previously received GA. The efficacy and safety evaluations were similar to Study 1 and the endpoints were broadly consistent, but the primary endpoint of Study 2 was the annualised relapse rate at 2 years, whereas the primary endpoint of Study 1 was the proportion of subjects relapsed at 2 years. Median age: 37 years, median years since diagnosis: 3.0 years and median EDSS score at baseline: 2.5. Patients were randomised to receive Tecfidera 240 mg twice a day (n=359), Tecfidera 240 mg three times a day (n=344), placebo (n=363) or glatiramer acetate (n=351) for up to 2 years. Median time on study was 96 weeks for all treatment groups.
The annualised relapse rate was significantly lower in patients treated with Tecfidera than in patients treated with placebo at 2 years. Secondary endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of T1 hypointense lesions, proportion of patients relapsed and time to confirmed disability progression defined as in Study 1.
Tecfidera had a clinically meaningful and statistically significant effect on the primary endpoint and secondary relapse and MRI endpoints. In Study 2, the annualised relapse rate for glatiramer acetate versus placebo was 0.286 and 0.401, corresponding to a reduction of 29% (p=0.013) which is consistent with approved product labelling. The results for this study are shown in Table 5.
 Pooled results at 2 years for Study 1 and Study 2 showed consistent and statistically significant results for Tecfidera versus placebo in all primary and secondary endpoints, including time to confirmed disability progression (32% relative reduction compared to placebo).
Pooled results at 2 years for Study 1 and Study 2 showed consistent and statistically significant results for Tecfidera versus placebo in all primary and secondary endpoints, including time to confirmed disability progression (32% relative reduction compared to placebo).
Study 3 (ENDORSE) enrolled eligible patients from Study 1 and 2 into an 8-year two phase extension study of 1736 patients with RRMS. The first phase was a multicenter, parallel-group, randomised, dose blind, dose comparison study in which patients received Tecfidera at a dose of 240 mg twice a day or 240 mg three times a day. The second phase was an open-label study during which all patients received Tecfidera at a dose of 240 mg twice a day. Eligible patients were enrolled at Week 96 (Visit 24) of their previous Study 1 or Study 2 visit, which served as the Baseline Visit for this extension study.
The primary objective of Study 3 was to evaluate the long-term safety of Tecfidera. The secondary objectives were to evaluate the long-term efficacy of Tecfidera using clinical endpoints (including relapse and ARR) and disability progression (EDSS) and on MS brain lesions on MRI scans.
The median age of patients was 40.0 years. Most patients (945 participants, 54%) were in the study for 7 years or longer and the median time spent in the study (min, max) was 6.759 (0.04, 10.98) years.
In the first year of treatment with Tecfidera in Study 3, the adjusted ARR (95% CI) ranged from 0.125 (0.084, 0.188) to 0.183 (0.108, 0.308), and remained low in the eighth year, ranging from 0.077 (0.039, 0.153) to 0.129 (0.063, 0.265), in all treatment arms. During the overall study period, the adjusted ARR (95% CI) ranged from 0.126 (0.098, 0.162) to 0.185 (0.129, 0.265) and the majority of patients treated with Tecfidera (between 59% and 69%) had no relapses. The estimated proportion of relapse (95% CI), at 8 years (384 weeks), in Study 3 ranged from 0.414 (0.314, 0.531) to 0.502 (0.426, 0.584).
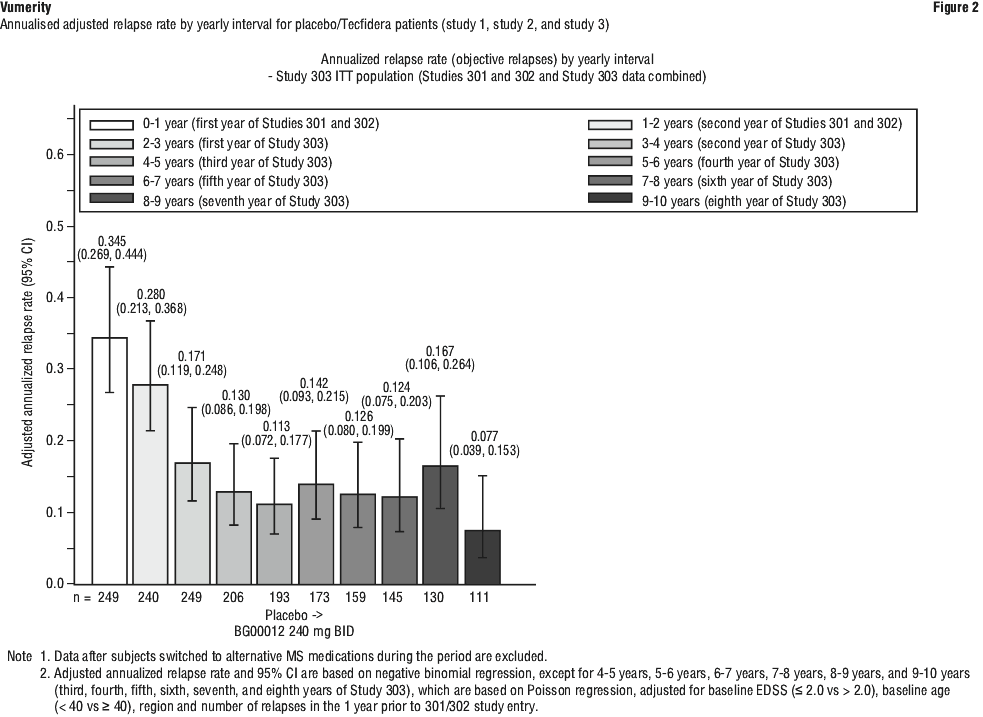
In an integrated analysis of Study 1 and Study 2 with Study 3, patients continuously treated with Tecfidera twice a day/ twice a day (n=501; patients treated with Tecfidera 240 mg twice a day in Study 1 or 2 and then Tecfidera 240 mg twice a day in Study 3), adjusted ARR was 0.187 (95% CI, 0.156, 0.224) in Study 1 and Study 2, and was 0.141 (95% CI, 0.119, 0.167) in Study 3. The data in Figure 1 demonstrate that the adjusted ARR in patients treated with Tecfidera was steady throughout the treatment time in Study 1 or 2 and Study 3. For placebo/ Tecfidera patients (n=249; patients treated with placebo in Study 1 or 2 and then switched to Tecfidera in Study 3), adjusted ARR was 0.330 (95% CI, 0.266, 0.408) and decreased after initiating Tecfidera in Study 3, to 0.149 (95% CI, 0.116, 0.190), shown in Figure 2.
In Study 3, the mean (median) EDSS score at baseline ranged from 2.37 (2.0) to 2.64 (2.0). The estimated proportion of patients with confirmed progression (95% CI) in the eighth year of Study 3 after treatment with Tecfidera ranged from 0.314 (0.268, 0.365) to 0.387 (0.311, 0.475).
In an integrated analysis of Study 1 and Study 2 with Study 3, at Week 480, the estimated proportion (95%) of patients with confirmed disability progression (Study 1, 2 and 3 data combined) was 0.349 (0.302, 0.401) in the Tecfidera twice a day/twice a day group and 0.362 (0.292, 0.443) in the placebo/Tecfidera group.
In Study 3, 752 patients were included in an MRI cohort, which included patients who had previously been included in the MRI cohort of Study 1 or Study 2. Due to sample size restrictions (by year 8 all groups had < 30 patients), MRI results are presented only through Year 6 of Study 3. Across all treatment arms, the adjusted mean number of new or newly enlarging T2 lesions relative to Study 3 baseline over 6 years ranged from 3.911 to 8.650 (the adjusted mean was based on negative binomial regression, adjusted for region and baseline volume of T2 lesions). The median number of new or newly enlarging T2 lesions over 6 years ranged from 1.0 to 3.0. Across all treatment arms, the mean (median) number of Gd+ lesions at 6 years ranged from 0.0 (0.0) to 0.7 (0.0). The percentage of patients with no Gd+ lesions ranged from 84% to 100%. The mean number of new T1 hypointense lesions over 6 years, adjusted for region and baseline volume of T1 lesions (based on negative binomial regression), ranged from 1.060 to 4.326. The median ranged from 1.0 to 2.5.

5.2 Pharmacokinetic Properties
Orally administered diroximel fumarate undergoes rapid presystemic hydrolysis by esterases and is converted to its primary metabolite, monomethyl fumarate, and an inactive metabolite HES. Diroximel fumarate is not quantifiable in plasma following oral administration of Vumerity. Therefore, all pharmacokinetic analyses related to Vumerity were performed with plasma monomethyl fumarate concentrations. Pharmacokinetic data were obtained in subjects with MS and healthy volunteers. Pharmacokinetic assessment showed that the pharmacokinetic parameters of monomethyl fumarate after administration of 462 mg Vumerity and 240 mg Tecfidera in adults are similar; therefore, Vumerity is expected to provide a similar overall efficacy and safety profile to Tecfidera.
Absorption.
The median time to reach maximum observed concentration of monomethyl fumarate is 2.5 to 3 hours. The peak plasma concentration (Cmax) and overall exposure (area under the concentration-time curve [AUC]) increased dose proportionally in the dose range studied (49 mg to 980 mg). Following administration of Vumerity 462 mg twice a day in MS patients (study EVOLVE-MS-1), the mean Cmax of monomethyl fumarate was 2.11 mg/L. The mean area under the concentration-time curve from time 0 to time of the last measurable concentration (AUClast) after a morning dose in MS patients was 4.15 mg*hr/L. The mean steady state daily AUC (AUCss) of monomethyl fumarate was estimated to be 8.32 mg*hr/L in MS patients.
Co-administration of Vumerity with a high-fat, high-calorie meal did not affect the AUC of monomethyl fumarate but resulted in an approximately 44% reduction in Cmax compared to fasted state. The monomethyl fumarate Cmax with low-fat and medium-fat meals was reduced by approximately 12% and 25%, respectively.
Distribution.
The apparent volume of distribution (Vd) for monomethyl fumarate is between 72 L and 83 L in healthy volunteers after administration of Vumerity. Human plasma protein binding of monomethyl fumarate was less than 25% and was not concentration dependent.
Metabolism.
In humans, Vumerity is extensively metabolised by esterases, which are ubiquitous in the GI tract, blood, and tissues, before it reaches the systemic circulation. Esterase metabolism of diroximel fumarate produces both monomethyl fumarate, the active metabolite, and HES, an inactive metabolite.
Further metabolism of monomethyl fumarate occurs through esterases followed by the TCA cycle, with no involvement of the CYP system. Fumaric and citric acid, and glucose are the major metabolites of monomethyl fumarate in plasma.
Excretion.
Monomethyl fumarate is mainly eliminated as carbon dioxide in the expired air with only trace amounts recovered in urine. The terminal half-life (t1/2) of monomethyl fumarate is approximately 1 hour, and no accumulation in monomethyl fumarate plasma exposures occurred with multiple doses of Vumerity. In a study with dimethyl fumarate, exhalation of CO2 was determined to be the primary route of elimination accounting for approximately 60% of the dose. Renal and fecal elimination are secondary routes of elimination, accounting for 15.5% and 0.9% of the dose, respectively.
HES is eliminated from plasma with a t1/2 of 10.7 hours to 14.8 hours. HES is mainly eliminated in urine.
Linearity.
Vumerity exposure increases in an approximately dose proportional manner with single and multiple doses in the 49 to 980 mg dose range studied.
Body weight, gender and age.
Body weight is the main covariate of exposure (by Cmax and AUC) of monomethyl fumarate after administration of Vumerity and Tecfidera, however it did not affect safety and efficacy measures evaluated in the clinical studies. Gender, age and race did not have a statistically significant impact on Cmax and AUC.
Paediatric.
The PK profile of monomethyl fumarate after administration of Vumerity has not been studied. The PK profile of monomethyl fumarate after administration of Vumerity is anticipated to be similar between adults and paediatric patients. The PK profile of monomethyl fumarate after administration of dimethyl fumarate 240 mg twice a day in paediatric patients with RRMS was evaluated in an open-label single arm study in patients aged 13-17 years (n=21). The PK of dimethyl fumarate in these patients was consistent with that previously observed in adult patients.
Race and ethnicity.
Race and ethnicity have no effect on the PK of monomethyl fumarate after administration of Tecfidera. Limited data suggest no effect of race and ethnicity on the PK of monomethyl fumarate after administration of Vumerity.
Renal impairment.
A single-dose clinical study investigating the effect of renal impairment on the PK of the Vumerity metabolites monomethyl fumarate and HES was conducted. The study included cohorts with mild, moderate, and severe renal impairment and a healthy cohort and found no clinically relevant changes in monomethyl fumarate exposure. HES exposure increased by 1.3-, 1.8-, and 2.7-fold with mild, moderate, and severe renal impairment, respectively. In EVOLVE-MS-1, there were 205 subjects with mild renal impairment (glomerular filtration rate < 90 mL/min/1.73 cm3 and ≥ 60 mL/min/1.73 cm3) and there were no clinically relevant differences in safety findings in this group as compared to subjects with no renal impairment. There are no data available on long-term use of Vumerity in patients with moderate or severe renal impairment.
Hepatic impairment.
As diroximel fumarate and monomethyl fumarate are metabolised by esterases, without the involvement of CYP450 system, hepatic impairment is not expected to affect exposure to monomethyl fumarate and HES. Therefore, no studies have been conducted in subjects with hepatic impairment and no dosage adjustment is necessary.
Alcohol.
Administration of Vumerity at the same time with 5% v/v and 40% v/v ethanol did not alter total monomethyl fumarate exposure relative to administration with water, demonstrating that the co-ingestion of ethanol does not induce dose dumping. The mean peak plasma monomethyl fumarate concentration for diroximel fumarate was decreased by 9% and 21%, when co-administered with 240 mL of 5% v/v and 40% v/v of ethanol, respectively.
5.3 Preclinical Safety Data
Genotoxicity.
Diroximel fumarate was not mutagenic in the in vitro bacterial reverse mutation assay. It was clastogenic in the in vitro chromosomal aberration assay in human peripheral blood lymphocytes, but not clastogenic/genotoxic in vivo in the rat micronucleus and comet assays. The active metabolite, monomethyl fumarate, was negative in the in vitro bacterial reverse mutation assay and in an in vivo rat bone marrow cytogenetic test but was clastogenic in an in vitro chromosomal aberration assay in human peripheral blood lymphocytes. The major inactive metabolite, HES, was not mutagenic in the in vitro bacterial reverse mutation assay and was not clastogenic in the in vitro chromosomal aberration assay in human peripheral blood lymphocytes. The weight of evidence does not suggest a risk of genotoxicity in patients, with negative results in in vivo studies considered more reliable than the inconsistent findings in in vitro studies.
Carcinogenicity.
Carcinogenicity studies of diroximel fumarate were conducted in transgenic rasH2 mice and in rats. Maximum exposures to MMF were low in rats and male mice (2x to 3x the maximum clinical AUC), and maximum exposures to HES were low in both species (both sexes, 1x to 4x). More adequate exposures to MMF were seen in female mice (exposure ratio 11) that received the limit dose. In transgenic rasH2 mice, diroximel fumarate was administered orally to males at doses of 30, 100, or 300 mg/kg/day and to females at doses of 30, 100, 300, or 1000 mg/kg/day for 26 weeks. Diroximel fumarate was not carcinogenic in either sex (monomethyl fumarate exposure was up to approximately 3x and 11x and HES exposure was up to approximately 1x and 4x in males and females, respectively, relative to exposure at the MRHD of diroximel fumarate on an AUC basis). In the rat study, diroximel fumarate was administered orally at doses of 15, 50, or 150 mg/kg/day to male and female animals for 90 and 94 weeks, respectively. Diroximel fumarate was not carcinogenic in female rats; in males, diroximel fumarate increased the incidence of testicular Leydig cell adenomas at 150 mg/kg/day (monomethyl fumarate and HES exposure was approximately 2x at the MRHD).
Leydig cell adenomas have been reported in male rats following treatment with dimethyl fumarate. The rat is particularly sensitive to developing this tumour type and the relevance of these findings to human risk is considered low.6 Pharmaceutical Particulars
6.1 List of Excipients
Capsule contents (enteric-coated minitablets).
Methacrylic acid-ethyl acrylate copolymer (1:1), crospovidone, microcrystalline cellulose, colloidal anhydrous silica, triethyl citrate, purified talc, magnesium stearate.
Capsule shell.
Hypromellose, titanium dioxide, potassium chloride, carrageenan, TekPrint SW-9008 Black Ink (capsule print - Proprietary Ingredient ID 2343).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
Store in original bottle to protect from moisture.
6.5 Nature and Contents of Container
HDPE bottle with a polypropylene lid and silica gel desiccant.
Pack-size of 120 capsules.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
C11H13NO6, 2-Butenedioic acid (2E)-, 1-[2-(2,5-dioxo-1-pyrrolidinyl)ethyl] 4-methyl ester, molecular mass 255.22 grams/mole.

CAS number.
The CAS Registry Number is 1577222-14-0.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
