What is in this leaflet
This leaflet answers some common questions about Vyndamax. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Vyndamax against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Vyndamax is used for
Your medicine, which contains tafamidis, is used to treat a disease called transthyretin amyloid cardiomyopathy, also known as ATTR-CM.
In patients with ATTR-CM, a protein called transthyretin (TTR) breaks up and may form fibrils called amyloid. Amyloid can build up between cells in your heart and in other places in your body, preventing your heart from working normally and causing symptoms.
Vyndamax is used to prevent TTR from breaking up and forming amyloid.
This medicine belongs to a group of medicines called transthyretin stabilisers.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
Vyndamax is believed to have no or negligible influence on the ability to drive and use machines.
This medicine is not recommended for use in children as ATTR-CM is not a condition common in children.
Before you take Vyndamax
When you must not take it
Do not take Vyndamax if you have an allergy to:
- any medicine containing tafamidis
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed if you are taking this medicine. The active ingredient in Vyndamax may pass into breast milk and there is a possibility that your baby may be affected.
Do not give this medicine to a child under the age of 18 years. Safety and effectiveness in children younger than 18 years have not been established.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have, or have had, any of the following medical conditions:
- an illness requiring an organ transplant
- a medical condition affected by excessive sorbitol
- severe liver problems
- severe kidney problems.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you are able to become pregnant, you must use birth control while you are taking Vyndamax and should continue using birth control for one month after stopping treatment with Vyndamax. There are no data on the use of Vyndamax in pregnant women.
If you have not told your doctor about any of the above, tell him/ her before you start taking Vyndamax.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Vyndamax may interfere with each other. These include:
- methotrexate, a medicine used to treat rheumatoid arthritis or some cancers
- rosuvastatin, a medicine used to treat high cholesterol
- imatinib, a medicine used for some cancer treatment.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take Vyndamax
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
Take one 61 mg capsule once a day.
One 61 mg Vyndamax capsule will produce the same level of active ingredient in your blood and give the same effect as four 20 mg capsules of Vyndaqel (total dose = 80 mg), even though one milligram of Vyndamax is not the same as one milligram of Vyndaqel.
Do not take Vyndamax and Vyndaqel at the same time.
How to take it
Swallow the capsule whole with a full glass of water. The capsule should not be crushed or cut.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine with or without food.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If it is within 6 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Vyndamax. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using Vyndamax
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Vyndamax.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not take Vyndamax to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how Vyndamax affects you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Vyndamax.
This medicine may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- feeling weak or a lack of energy
- feeling unbalanced when standing or walking
- fall
- sinusitis
- cataract (clouding of the lens in your eye)
- flatulence
- muscle or joint pain
- skin ulcer
- bladder infection
- excessive sweating.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
After using Vyndamax
Storage
Keep your capsules in the pack until it is time to take them. If you take the capsules out of the pack they may not keep well.
Keep your capsules in a cool dry place where the temperature stays below 25°C.
Do not store Vyndamax or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Vyndamax 61 mg is a reddish brown, opaque, oblong capsule printed with "VYN 61" in white.
They are supplied in blister packs of 30 capsules.
Ingredients
Vyndamax contains 61 mg of tafamidis as the active ingredient.
- Butylated hydroxytoluene
- Ethanol
- Gelatin
- Glycerol
- Iron oxide red
- Isopropyl alcohol
- Polysorbate 20
- Polyvinyl acetate phthalate
- Povidone
- Propylene glycol
- Macrogol 400
- Mannitol
- Sorbitol
- Strong ammonium solution
- Titanium dioxide.
This medicine does contain sulfites.
This medicine does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Supplier
Vyndamax is supplied in Australia by:
Pfizer Australia Pty Ltd
Sydney, NSW
Toll Free Number: 1800 675 229
www.pfizer.com.au
AUST R 314813
This leaflet was prepared in March 2020.
® Registered Trademark
© Pfizer Australia Pty Ltd 2020
Published by MIMS December 2020
 The treatment-emergent adverse events with a greater incidence in patients treated with tafamidis meglumine (pooled tafamidis meglumine 20 mg + 80 mg) than placebo in the pivotal phase 3 Study B3461028 are shown in Table 2.
The treatment-emergent adverse events with a greater incidence in patients treated with tafamidis meglumine (pooled tafamidis meglumine 20 mg + 80 mg) than placebo in the pivotal phase 3 Study B3461028 are shown in Table 2. A lower proportion of tafamidis meglumine treated patients compared to placebo discontinued due to an adverse event in the 30-month placebo-controlled trial in patients diagnosed with ATTR-CM [40 (22.7%), 16 (18.2%), and 51 (28.8%) from the tafamidis meglumine 80 mg (administered as four 20 mg capsules), tafamidis meglumine 20 mg, and placebo groups, respectively].
A lower proportion of tafamidis meglumine treated patients compared to placebo discontinued due to an adverse event in the 30-month placebo-controlled trial in patients diagnosed with ATTR-CM [40 (22.7%), 16 (18.2%), and 51 (28.8%) from the tafamidis meglumine 80 mg (administered as four 20 mg capsules), tafamidis meglumine 20 mg, and placebo groups, respectively]. The primary analysis used the Finkelstein-Schoenfeld (F-S) method to perform a hierarchical combination of all-cause mortality and frequency of cardiovascular-related hospitalisations, which is defined as the number of times a subject is hospitalised (i.e. admitted to a hospital) for cardiovascular-related morbidity. The F-S method compared each patient to every other patient within each stratum in a pair-wise manner to determine which member of the pair had experienced a more favourable outcome (i.e. "won"), proceeding in a hierarchical fashion using all-cause mortality followed by frequency of cardiovascular-related hospitalisations when patients could not be differentiated based on mortality.
The primary analysis used the Finkelstein-Schoenfeld (F-S) method to perform a hierarchical combination of all-cause mortality and frequency of cardiovascular-related hospitalisations, which is defined as the number of times a subject is hospitalised (i.e. admitted to a hospital) for cardiovascular-related morbidity. The F-S method compared each patient to every other patient within each stratum in a pair-wise manner to determine which member of the pair had experienced a more favourable outcome (i.e. "won"), proceeding in a hierarchical fashion using all-cause mortality followed by frequency of cardiovascular-related hospitalisations when patients could not be differentiated based on mortality. Analysis of the individual components of the primary analysis (all-cause mortality and cardiovascular-related hospitalisation) also demonstrated significant reductions for tafamidis meglumine versus placebo.
Analysis of the individual components of the primary analysis (all-cause mortality and cardiovascular-related hospitalisation) also demonstrated significant reductions for tafamidis meglumine versus placebo.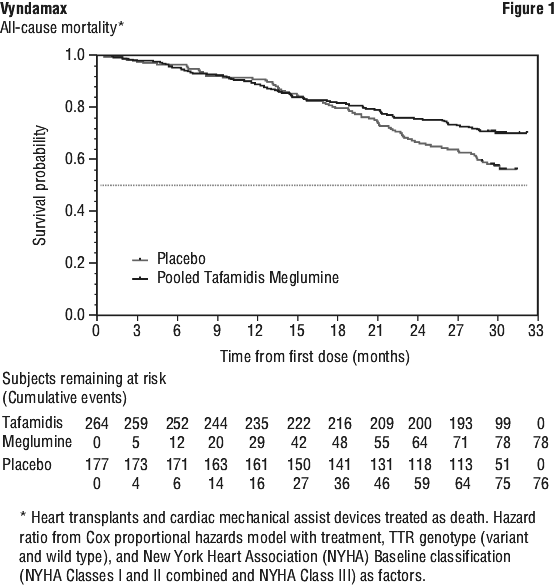 There were significantly fewer cardiovascular-related hospitalisations with tafamidis meglumine compared with placebo with a reduction in risk of 32.4% (Table 5).
There were significantly fewer cardiovascular-related hospitalisations with tafamidis meglumine compared with placebo with a reduction in risk of 32.4% (Table 5). The treatment effect of tafamidis meglumine on functional capacity and health status was assessed by the 6-Minute Walk Test (6MWT) and the Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS) score, respectively. A significant treatment effect favouring tafamidis meglumine was first observed at Month 6 and remained consistent through Month 30 on both 6MWT distance and KCCQ-OS score (Figure 2 and Table 6).
The treatment effect of tafamidis meglumine on functional capacity and health status was assessed by the 6-Minute Walk Test (6MWT) and the Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS) score, respectively. A significant treatment effect favouring tafamidis meglumine was first observed at Month 6 and remained consistent through Month 30 on both 6MWT distance and KCCQ-OS score (Figure 2 and Table 6). The KCCQ-OS score is composed of four domains including Total Symptom (Symptom Frequency and Symptom Burden), Physical Limitation, Quality of Life, and Social Limitation. All four domains favoured tafamidis meglumine compared to placebo at Month 30 (Figure 2 and Table 6). KCCQ-OS and domain scores range from 0-100 with higher scores representing better health status. The cumulative distribution and distribution for change from Baseline to Month 30 for KCCQ-OS (Figure 3) show that the proportion of patients with declining KCCQ-OS scores was lower for the pooled tafamidis meglumine treated group compared to placebo (Figure 3).
The KCCQ-OS score is composed of four domains including Total Symptom (Symptom Frequency and Symptom Burden), Physical Limitation, Quality of Life, and Social Limitation. All four domains favoured tafamidis meglumine compared to placebo at Month 30 (Figure 2 and Table 6). KCCQ-OS and domain scores range from 0-100 with higher scores representing better health status. The cumulative distribution and distribution for change from Baseline to Month 30 for KCCQ-OS (Figure 3) show that the proportion of patients with declining KCCQ-OS scores was lower for the pooled tafamidis meglumine treated group compared to placebo (Figure 3).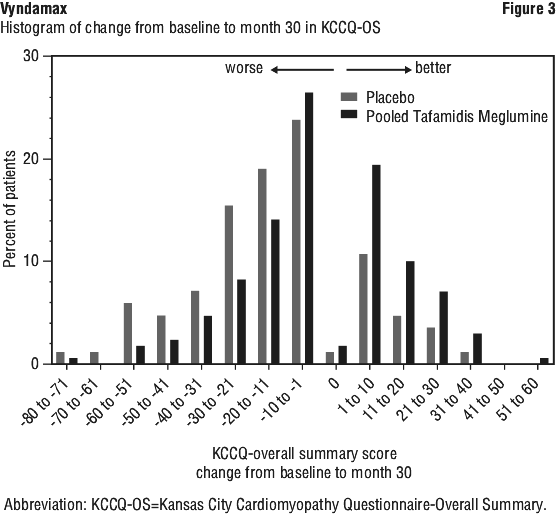
 In the pivotal study, patients who exhibited > 32% TTR stabilisation were considered stabilised. At Month 1, a significantly greater proportion of patients in the pooled tafamidis meglumine group (211/245 [86.1%] patients) demonstrated TTR stabilisation than was observed for patients in the placebo group (6/170 [3.5%] patients) (p < 0.0001).
In the pivotal study, patients who exhibited > 32% TTR stabilisation were considered stabilised. At Month 1, a significantly greater proportion of patients in the pooled tafamidis meglumine group (211/245 [86.1%] patients) demonstrated TTR stabilisation than was observed for patients in the placebo group (6/170 [3.5%] patients) (p < 0.0001). Results of the primary analysis, 6MWT at Month 30 and KCCQ-OS at Month 30 were statistically significant for both the 80 mg and 20 mg doses of tafamidis meglumine vs. placebo, with similar results for both doses.
Results of the primary analysis, 6MWT at Month 30 and KCCQ-OS at Month 30 were statistically significant for both the 80 mg and 20 mg doses of tafamidis meglumine vs. placebo, with similar results for both doses.

