1 Name of Medicine
Belzutifan.
2 Qualitative and Quantitative Composition
Each Welireg tablet contains 40 mg of belzutifan.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Welireg 40 mg tablet is a blue oval shaped, film-coated tablet debossed with "177" on one side and plain on the other side.
4.1 Therapeutic Indications
Von Hippel-Lindau (VHL) disease associated tumours.
Welireg (belzutifan) is indicated for the treatment of adult patients with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma (RCC), central nervous system (CNS) haemangioblastomas, or pancreatic neuroendocrine tumours (pNET), not requiring immediate surgery.
Advanced renal cell carcinoma (RCC).
Welireg (belzutifan) is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) following a programmed cell death 1/ligand 1 (PD-1/L1) and a vascular endothelial growth factor (VEGF)-targeted therapy.4.2 Dose and Method of Administration
The recommended dose of Welireg is 120 mg (three 40 mg tablets) administered orally once daily, with or without food. Swallow tablets whole.
If a dose of Welireg is missed, it can be taken as soon as possible on the same day. Resume the regular daily dose schedule for Welireg the next day. Extra tablets should not be taken to make up for the missed dose. If vomiting occurs any time after taking Welireg, do not retake the dose. The next dose should be taken the next day. Treatment should continue until disease progression or unacceptable toxicity occurs.
Dose modifications.
Dosage modifications for Welireg for adverse reactions are summarised in Table 1.

Paediatric patients.
Safety and efficacy of Welireg have not been established in paediatric patients less than 18 years of age (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Geriatric patients.
No dose adjustment of Welireg is recommended in elderly patients (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Renal impairment.
No dose adjustment of Welireg is recommended in patients with mild and moderate renal impairment. Welireg has not been studied in patients with severe renal impairment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No dose adjustment of Welireg is recommended in patients with mild hepatic impairment. Welireg has not been studied in patients with moderate or severe hepatic impairment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).4.3 Contraindications
None.
4.4 Special Warnings and Precautions for Use
Anaemia due to decreased erythropoietin.
Welireg can cause severe anaemia that can require blood transfusion.
In a clinical trial (LITESPARK-004) with Welireg for the treatment of patients with VHL disease-associated RCC, anaemia was reported in 55 patients (90.2%). Grade 3 anaemia occurred in 6 patients (9.8%) (see Section 4.8 Adverse Effects (Undesirable Effects)). Median time to onset of all Grade anaemia events was 30 days (range: 1 day to 8.38 months). Of the 13 patients that were treated with an erythropoiesis-stimulating agent (ESA), 4 received treatment with both an ESA and blood transfusions, while 9 received treatment with an ESA alone. Patients received an ESA based on haemoglobin levels and physician discretion (see Section 5.1 Pharmacodynamic Properties). In LITESPARK-005, a clinical trial with Welireg for the treatment of patients with advanced RCC, anaemia occurred in 83% of patients, 119 patients (32%) had Grade 3 and 2 patients (0.5%) had Grade 4 anaemia (see Section 4.8 Adverse Effects (Undesirable Effects)). Median time to onset of anaemia was 29 days (range: 1 day to 27 months). Of the patients with anaemia, 67 patients (22%) received transfusions only, 62 patients (20%) of patients received ESAs only and 42 patients (14%) received both transfusion and ESAs. The median number of ESA doses administered to patients was 6.5 (range: 1-87). Patients received an ESA based on haemoglobin levels and physician discretion. In another clinical trial (Study-001) for the treatment of non-VHL disease-associated advanced solid tumours using the same dose of Welireg, anaemia was reported in 44 patients (75.9%). Grade 3 anaemia occurred in 16 patients (27.6%).
Monitor for anaemia before initiation of and periodically throughout treatment with Welireg. Closely monitor patients who are dual UGT2B17 and CYP2C19 poor metabolisers due to potential increases in exposure that may increase the incidence or severity of anaemia. For patients who develop Grade 3 anaemia, withhold Welireg and treat according to standard medical practice, including erythropoiesis-stimulating agent (ESA) administration and/or transfusion until resolved to ≤ Grade 2. For recurrent Grade 3 anaemia, consider discontinuing Welireg. For patients who develop Grade 4 anaemia, withhold Welireg; then resume at a reduced dose or permanently discontinue for recurrent Grade 4 anaemia (see Section 4.2 Dose and Method of Administration).
Hypoxia.
Welireg can cause severe hypoxia that may require discontinuation, supplemental oxygen, or hospitalisation.
In a clinical trial (LITESPARK-004) with Welireg for the treatment of patients with VHL disease-associated RCC, Grade 3 hypoxia occurred in 1 patient (1.6%) (see Section 4.8 Adverse Effects (Undesirable Effects)). In LITESPARK-005, a clinical trial with Welireg for the treatment of patients with advanced RCC, hypoxia occurred in 15% of patients and 38 patients (10%) had Grade 3 hypoxia and 1 patient (0.3%) had Grade 4 hypoxia (see Section 4.8 Adverse Effects (Undesirable Effects)). Of the patients with hypoxia, 70% were treated with oxygen therapy. Median time to onset of hypoxia was 1 month (range: 1 day to 21 months).
In another clinical trial (Study-001) for the treatment of non-VHL disease-associated advanced solid tumours using the same dose of Welireg, hypoxia occurred in 17 patients (29.3%), Grade 3 hypoxia occurred in 9 patients (15.5%).
Monitor oxygen saturation with pulse oximetry before initiation of and periodically throughout treatment with Welireg. For Grade 3 asymptomatic hypoxia (e.g. pulse oximetry < 88% or PaO2 ≤ 55 mmHg at rest), consider providing supplemental oxygen and consider continuing or withholding treatment. If withheld, resume at a reduced dose. For patients who have Grade 3 symptomatic hypoxia (e.g. pulse oximetry < 88% or PaO2 ≤ 55 mmHg at rest), withhold Welireg, treat hypoxia, and consider dose reduction. If symptomatic hypoxia continues to recur, discontinue treatment. For Grade 4 hypoxia (life-threatening airway compromise; urgent intervention indicated e.g. tracheotomy or intubation), permanently discontinue treatment (see Section 4.2 Dose and Method of Administration).
Advise patients to report signs and symptoms of hypoxia immediately to a healthcare provider.
Embryo-fetal toxicity.
Based on findings in animals, Welireg may cause fetal harm, including fetal loss, in humans. In a rat study, belzutifan caused embryo-fetal toxicity when administered during the period of organogenesis at maternal exposures that were lower than the human exposures at the recommended dose of 120 mg daily. Advise females of reproductive potential to use effective non-hormonal contraceptive during treatment with Welireg and for 1 week after the last dose due to the potential risk to the fetus. This is because the use of Welireg may reduce the efficacy of hormonal contraceptives. Advise male patients with female partners of reproductive potential to use highly effective contraception during treatment with Welireg and for 1 week after the last dose (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 4.6 Fertility, Pregnancy and Lactation).
Dual UGT2BI7 and CYP2C19 poor metabolisers.
Patients who are dual UGT2B17 and CYP2C19 poor metabolisers have higher belzutifan exposures, which may increase the incidence and severity of adverse reactions of belzutifan and should be closely monitored (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties).
Use in hepatic impairment.
No dose adjustment of Welireg is recommended in patients with mild hepatic impairment. Welireg has not been studied in patients with moderate or severe hepatic impairment (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
No dose adjustment of Welireg is recommended in patients with mild or moderate renal impairment. Welireg has not been studied in patients with severe renal impairment (see Section 4.2 Dose and Method of Administration; Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
No dosage adjustment of Welireg is recommended in geriatric patients. Of the 61 patients with VHL disease-associated RCC (LITESPARK-004) treated with Welireg, only 2 patients were 65 years and over. Of the patients with advanced RCC who received Welireg in LITESPARK-005, 38% (142 patients) were ≥ 65 years old (see Section 5.1 Pharmacodynamic Properties, Clinical trials). Another clinical trial (Study-001) for the treatment of non-VHL disease-associated advanced solid tumours included 24 patients were 65 years and over. No overall difference in safety or efficacy was reported between patients who were 65 years and over and younger patients (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 5.2 Pharmacokinetic Properties).
Paediatric use.
Safety and effectiveness of Welireg in paediatric patients under 18 years of age have not been established.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
In vitro and pharmacogenomic studies indicate that belzutifan is metabolised by UGT2B17 and by CYP2C19.
Effects of Welireg on other drugs.
In vitro studies have shown that belzutifan induces CYP3A4.
Co-administration of Welireg with CYP3A4 substrates, including hormonal contraceptives, decreases concentrations of CYP3A substrates, which may reduce the efficacy of these substrates. The magnitude of this decrease may be more pronounced in patients who are dual UGT2B17 and CYP2C19 poor metabolisers (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 5.2 Pharmacokinetic Properties).
Co-administration of Welireg with hormonal contraceptives may lead to contraceptive failure or an increase in breakthrough bleeding.
Belzutifan inhibits MATE2K (IC50 0.7 microM), OCT1 (IC50 47 microM) and MATE1 (IC50 39 microM) in vitro while the clinical unbound Cmax is 1.8 microM. Welireg might increase plasma exposures of co-administered drugs that are predominantly cleared by these transporters.
Effects of other drugs on Welireg.
Co-administration with inhibitors of UGT2B17 or CYP2C19 is expected to increase plasma belzutifan exposure. Dose adjustment is not required on co-administration with inhibitors of UGT2B17 or CYP2C19. Drugs that induce CYP2C19 are expected to reduce plasma exposures of belzutifan.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No fertility studies in animals were conducted. Testicular atrophy and hypospermia, associated with decreased sperm count and motility and abnormal sperm morphology, were observed in general toxicology studies in rats at exposures lower than the human exposure at the recommended dose of 120 mg daily, and did not reverse at the end of a 26-week recovery period. Belzutifan caused post implantation loss in pregnant rats at exposures lower than the human exposure at the recommended dose of 120 mg daily. Based on findings in animals, Welireg may impair fertility in males and females of reproductive potential. Advise patients of this potential risk.
(Category D)
Based on findings in animal studies, Welireg may cause fetal harm, including fetal loss, when administered to a pregnant woman. There are no available data on the use of Welireg in pregnant women to evaluate drug-associated risk. In a rat embryo-fetal development study, administration of belzutifan during organogenesis caused embryo-fetal lethality, reduced fetal body weight, and fetal skeletal abnormalities at exposures similar to or below the human exposure at the recommended dose of 120 mg daily. Advise females of reproductive potential of the potential risk to a fetus.
There are no data on the presence of belzutifan or its metabolites in human milk, their effects on the breastfed child, or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with Welireg and for 1 week after the last dose.
Females and males of reproductive potential.
Pregnancy testing. Verify the pregnancy status of females of reproductive potential prior to initiating treatment with Welireg.
Contraception. Welireg may cause embryo-fetal harm, including fetal loss, when administered to a pregnant woman (see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy).
Females.
Advise females of reproductive potential to use highly effective contraception during treatment with Welireg and for at least 1 week after the last dose. Use of Welireg may reduce the efficacy of hormonal contraceptives. Advise patients using hormonal contraceptives to use an alternative non-hormonal contraceptive method or add a barrier method of contraception (e.g. male condom) during treatment with Welireg (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Males.
Advise male patients with female partners of reproductive potential to use highly effective contraception during treatment with Welireg and for at least 1 week after the last dose.4.7 Effects on Ability to Drive and Use Machines
Dizziness and fatigue may occur following administration of belzutifan (see Section 4.8 Adverse Effects (Undesirable Effects)).
Patients should be advised not to drive and use machines, until they are reasonably certain belzutifan therapy does not affect them adversely.
4.8 Adverse Effects (Undesirable Effects)
Clinical trials experience.
von Hippel-Lindau (VHL) disease associated tumours. Clinical trials experience.
The safety of Welireg was evaluated in an open-label Phase 2 clinical trial (LITESPARK-004), in 61 patients with VHL disease who had at least one measurable solid tumour localised to the kidney as defined by Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST v1.1) and who did not require immediate surgery. Patients were treated with Welireg 120 mg once daily (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The median duration of exposure to Welireg was 125 weeks (range: 8.4 to 163.1 weeks).
Adverse events.
Welireg was discontinued due to adverse events in 3 patients (4.9%): 1 patient for Grade 1 dizziness; one patient for haemorrhage intracranial and 1 patient due to death from a concomitantly administered toxic agent. Adverse events leading to dose interruption of Welireg occurred in 27 patients (44.3%) and adverse events leading to dose reduction of Welireg occurred in 9 patients (14.8%). The most common (≥ 10%) adverse event resulting in dose interruption or reduction of Welireg was fatigue. Serious adverse events occurred in 14 patients (23.0%), of which 4 were determined to be drug-related (anaemia, urinary tract infection, haemorrhage intracranial, and hypoxia).
The most common adverse events (≥ 25%) that occurred in patients treated with Welireg were anaemia, fatigue, headache, dizziness, and nausea. Table 2 summarises the adverse events reported in patients treated with Welireg.

Adverse drug reactions.
The most common adverse reactions under treatment with belzutifan were anaemia (90%), fatigue (70%), dizziness (44%) and nausea (36%).
The most common adverse reactions resulting in dose interruption of belzutifan were fatigue (13.1%), nausea (8.2%) and anaemia (4.9%). The most common adverse reactions resulting in dose reduction of belzutifan were fatigue (8.2%), anaemia (1.6%) and hypoxia (1.6%). Belzutifan was discontinued due to adverse reaction in 1 patient (1.6%) (Grade 1 dizziness).
Tabulated list of adverse reactions.
Adverse reactions reported in clinical studies with belzutifan are listed in Table 3 by MedDRA system organ class and by frequency. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), and very rare (< 1/10,000).

Advanced renal cell carcinoma.
The safety of belzutifan was evaluated in a Phase 3 clinical study (LITESPARK-005), in 372 patients with advanced RCC. Patients were treated with 120 mg belzutifan once daily. The median duration of exposure to belzutifan was 7.6 months (range 0.1 to 35.8 months).
The most common adverse reactions under treatment with belzutifan were anaemia (83%), fatigue (31%), dyspnoea (15%), hypoxia (15%), nausea (18%) and dizziness (12%).
The most common adverse reactions resulting in dose interruption of belzutifan were anaemia (8.6%), hypoxia (5.6%), fatigue (1.6%), dizziness (1.6%), dyspnoea (1.6%) and nausea (1.3%). The most common adverse reactions resulting in dose reduction of belzutifan were hypoxia (5.6%) and anaemia (3.0%). Belzutifan was discontinued due to adverse reaction in 5.9% of patients.
Tabulated list of adverse reactions.
Adverse reactions observed in clinical studies of belzutifan are listed in Table 4. These reactions are presented by system organ class and by frequency. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), and very rare (< 1/10,000).
 The safety of Welireg was also evaluated in a Phase 1 clinical trial (Study-001), in 58 patients with non-VHL disease-associated advanced solid tumours, treated with Welireg 120 mg once daily. Study-001 patients differed from VHL-associated RCC patients (LITESPARK-004). Study-001 patients were older, had worse ECOG PS, had metastatic disease, had prior therapies, had more comorbidities, and had lower baseline haemoglobin levels at treatment initiation. Study-001 had a median duration of exposure to Welireg of 25.4 weeks (range: 1.1 to 145.9 weeks). Welireg was discontinued due to adverse reactions in 6 patients (10.3%), most commonly due to hypoxia. Adverse reactions leading to dose interruption occurred in 24 patients (41.4%) and adverse reactions leading to dose reduction occurred in 6 patients (10.3%). The most common (≥ 10%) adverse reaction resulting in dose interruption or reduction was hypoxia. The most common adverse reactions (≥ 25%) that occurred in patients treated with Welireg were anaemia, fatigue, dyspnoea, nausea, hypoxia, cough, vomiting, and peripheral oedema.
The safety of Welireg was also evaluated in a Phase 1 clinical trial (Study-001), in 58 patients with non-VHL disease-associated advanced solid tumours, treated with Welireg 120 mg once daily. Study-001 patients differed from VHL-associated RCC patients (LITESPARK-004). Study-001 patients were older, had worse ECOG PS, had metastatic disease, had prior therapies, had more comorbidities, and had lower baseline haemoglobin levels at treatment initiation. Study-001 had a median duration of exposure to Welireg of 25.4 weeks (range: 1.1 to 145.9 weeks). Welireg was discontinued due to adverse reactions in 6 patients (10.3%), most commonly due to hypoxia. Adverse reactions leading to dose interruption occurred in 24 patients (41.4%) and adverse reactions leading to dose reduction occurred in 6 patients (10.3%). The most common (≥ 10%) adverse reaction resulting in dose interruption or reduction was hypoxia. The most common adverse reactions (≥ 25%) that occurred in patients treated with Welireg were anaemia, fatigue, dyspnoea, nausea, hypoxia, cough, vomiting, and peripheral oedema.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
There is no specific treatment for Welireg overdose. In cases of suspected overdose, if necessary, consider withholding Welireg and instituting supportive care. The highest dose of Welireg studied clinically was 240 mg total daily dose (120 mg twice a day or 240 mg once a day). Grade 3 hypoxia occurred at 120 mg twice a day and Grade 4 thrombocytopenia occurred at 240 mg once daily.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
Pharmacotherapeutic group: other antineoplastic agents, ATC code: L01XX74.
5.1 Pharmacodynamic Properties
Mechanism of action.
Belzutifan is an inhibitor of hypoxia-inducible factor 2 alpha (HIF-2α). HIF-2α is a transcription factor that plays a role in oxygen sensing by regulating genes that promote adaptation to hypoxia. Under normal oxygen levels, HIF-2α is targeted for ubiquitin-proteasomal degradation by VHL protein. Lack of functional VHL protein results in stabilisation and accumulation of HIF-2α. Upon stabilisation, HIF-2α translocates into the nucleus and interacts with hypoxia-inducible factor 1 beta (HIF-1β) domains to form a transcriptional complex that induces expression of downstream genes, including genes associated with cellular proliferation, angiogenesis, and tumour growth. Belzutifan binds to HIF-2α, and in conditions of hypoxia or impairment of VHL protein function, belzutifan blocks the HIF-2α-HIF-1β interaction, leading to reduced transcription and expression of HIF-2α target genes. In vivo, belzutifan has demonstrated anti-tumour activity in mouse xenograft models of clear cell renal cell carcinoma (ccRCC).
Circulating plasma levels of erythropoietin (EPO) were monitored in patients as a pharmacodynamic marker of HIF-2α inhibition. Reductions in EPO were observed to be dose/exposure dependent and showed a plateauing effect on reduction at exposures achieved with doses above 120 mg once daily. The maximum EPO suppression occurred following 2 weeks of consecutive dosing of Welireg (mean percent decrease from baseline of approximately 60%). Mean EPO levels gradually returned to baseline values after 12 weeks of treatment.
The incidence of Grade 3 anaemia increased with higher belzutifan exposure in patients with baseline haemoglobin levels < 12 g/dL (see Section 4.4 Special Warnings and Precautions for Use).
Cardiac electrophysiology.
At the recommended dose (120 mg once daily) for Welireg, there were no clinically relevant effects on the QTc interval.
Pharmacogenomics.
Belzutifan is primarily metabolised by UGT2B17 and CYP2C19. The activity of these enzymes varies among individuals who carry different genetic variants, which may impact belzutifan concentrations. Poor metabolisers are individuals who are considered to have no enzyme activity. Approximately 15% of Caucasians, 11% of Latinos, 6% of African Americans, 38% of South Asians, and 70% of East Asians are UGT2B17 poor metabolisers. Approximately 2% of Caucasians, 1% of Latinos, 5% of African Americans, 8% of South Asians, and 13% of East Asians are CYP2C19 poor metabolisers. Approximately 0.3% of Caucasians, 0.1% of Latinos, 0.3% of African Americans, 3% of South Asians, and 9% of East Asians are dual UGT2B17 and CYP2C19 poor metabolisers. Expected frequencies in the Japanese population for the UGT2B17, CYP2C19, and dual UGT2B17 and CYP2C19 poor metabolisers are approximately 77%, 19%, and 15%, respectively. Expected frequencies in the United States population for the UGT2B17, CYP2C19, and dual UGT2B17 and CYP2C19 poor metabolisers are approximately 16%, 3%, and 0.5%, respectively based on the reported proportion of the US population represented by major racial/ethnic groups.
The impact of CYP2C19 and UGT2B17 poor metabolisers on Welireg exposure was assessed in a population PK analysis. Based on the population PK model patients who are CYP2C19, UGT2B17, or dual UGT2B17 and CYP2C19 poor metabolisers, are projected to have 1.3-, 2.7-, or 3.3-fold the exposures (steady state AUC0-24hr), respectively, compared to a typical reference patient (UGT2B17 extensive metaboliser, CYP2C19 extensive/intermediate metaboliser) for the recommended dose. No dose adjustment is recommended based on exposure-response analyses for efficacy and safety and the risk-benefit profile.
Clinical trials.
Clinical studies in adult patients with von Hippel-Lindau (VHL) disease associated tumours.
The efficacy of Welireg was investigated in LITESPARK-004, an open-label Phase 2 clinical trial in 61 patients with VHL disease who had at least one measurable solid tumour (as defined by RECIST v1.1) localised to the kidney and who did not require immediate surgery. Patients received Welireg at a dose of 120 mg once daily. Patients were evaluated radiologically approximately 12 weeks after initiation of treatment and every 12 weeks thereafter. Treatment was continued until progression of disease or unacceptable toxicity. The study excluded patients who had any evidence of metastatic disease, either RCC or other VHL disease-associated tumours, an immediate need for surgical intervention for tumour treatment, any major surgical procedure completed within 4 weeks prior to study enrollment, any major cardiovascular event within 6 months prior to study drug administration, or prior systemic treatments for VHL disease-associated RCC.
The study population characteristics were: median age of 41 years, 3.3% age 65 or over; 52.5% male; 90.2% White; and 82.0% had an ECOG PS of 0 and 16.4% had an ECOG PS of 1. Seventy-seven percent of patients had prior RCC surgical procedures. Other VHL disease-associated tumours in patients included pancreatic lesions (100.0%) of which 36.1% were pancreatic neuroendocrine tumours, CNS haemangioblastomas (82.0%), and retinal angiomas (19.7%).
The primary efficacy endpoint for the treatment of VHL disease-associated RCC was objective response rate (ORR) measured by Integrated Radiology and Oncology Assessment (IRO) assessment using RECIST v1.1 as assessed by a central independent review committee (IRC). Additional efficacy endpoints included disease control rate (DCR), response duration, time to response (TTR), and time to surgery (TTS). Radiographic endpoints were assessed by IRC using RECIST v1.1. The clinical benefit of Welireg in reducing RCC tumour size and slowing the growth of tumours was supported by pre-treatment and post-treatment linear growth rate of 3.52 and 3.37 mm/year respectively in LITESPARK-004. A total of 91.8% of participants (56/61) had a decrease in the sum of target tumour diameters, including 3.3% (2/61) achieved complete response (see Figure 1). After a median follow-up time of 29.2 months, one out of 61 (2%) patients required an RCC tumour reduction procedure during treatment. In a natural history study, RCC patients undergoing active surveillance have demonstrated linear tumour growth of 3.56 mm/year and showed that 28.7% of patients had their first tumour reduction procedure within 24 months of follow-up. Table 5 summarises the efficacy results for VHL disease-associated RCC tumours in LITESPARK-004.

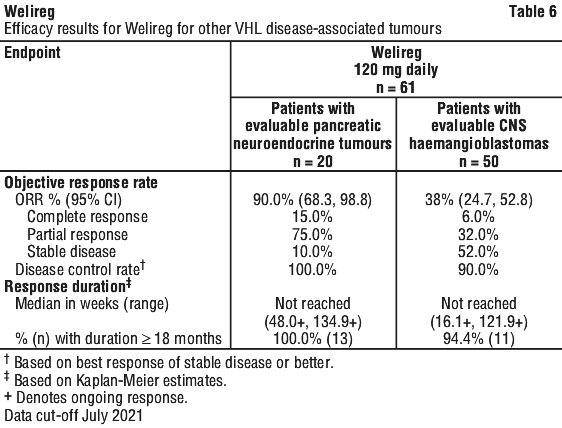
 Efficacy endpoints for the treatment of other VHL disease-associated tumours included ORR, DCR and response duration, as assessed by IRC using RECIST v1.1. These results are shown in Table 6.
Efficacy endpoints for the treatment of other VHL disease-associated tumours included ORR, DCR and response duration, as assessed by IRC using RECIST v1.1. These results are shown in Table 6.
Clinical studies in adult patients with advanced renal cell carcinoma (RCC).
The efficacy of belzutifan was evaluated in LITESPARK-005, an open-label, randomised, active-controlled Phase 3 clinical study comparing belzutifan with everolimus in 746 patients with unresectable, locally advanced or metastatic clear cell RCC that has progressed following PD-1/L1 checkpoint inhibitor and VEGF receptor targeted therapies either in sequence or in combination. Patients could have received up to 3 prior treatment regimens and must have measurable disease per RECIST v1.1. Patients were randomised in a 1:1 ratio to receive 120 mg belzutifan or 10 mg everolimus by oral administration once daily. Randomisation was stratified by International Metastatic RCC Database Consortium (IMDC) risk categories (favourable versus intermediate versus poor) and number of prior VEGF receptor targeted therapies (1 versus 2-3).
Patients were evaluated radiologically at Week 9 from the date of randomisation, then every 8 weeks through Week 49, and every 12 weeks thereafter.
Among the 746 patients in LITESPARK-005, the baseline characteristics were: median age 63 years (range 22-90 years), 42% age 65 or older; 78% male; 79% White; 12% Asian; 1.1% Black or African American; 43% ECOG performance status 0 and 55% ECOG performance status 1. Prior therapies: 13% of patients had 1 prior line of therapy, 43% had 2 prior lines of therapy and 43% had 3 prior lines of therapy; 49% received 2 to 3 prior VEGF receptor targeted therapies. Patient distribution by IMDC risk categories was 22% favourable, 66% intermediate, and 12% poor.
The primary efficacy outcome measures were Progression-Free Survival (PFS) measured by BICR using RECIST v1.1, and Overall Survival (OS). Secondary efficacy outcome measures included objective response rate (ORR), and duration of response (DOR) by BICR using RECIST v1.1.
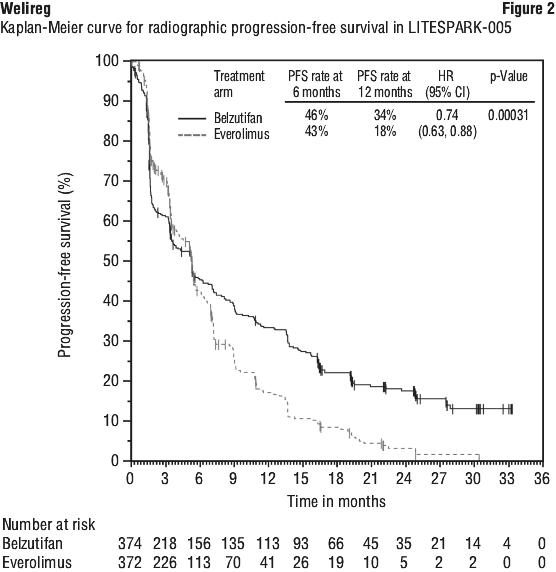
The trial demonstrated a statistically significant improvement of PFS for patients randomised to Welireg compared with everolimus. The efficacy results for advanced RCC in LITESPARK-005 are summarised in Table 7.
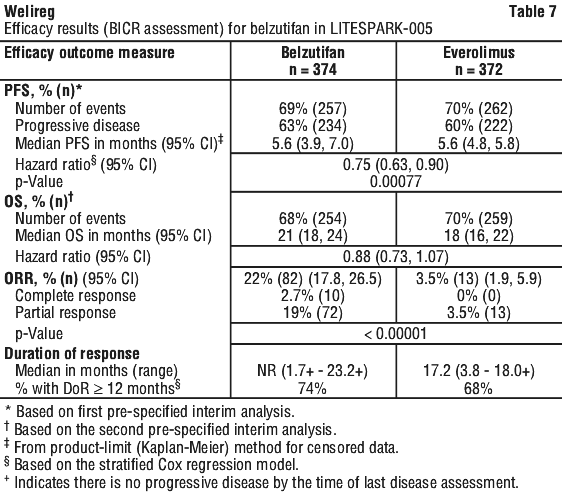 At a subsequent pre-specified analysis with median follow-up time of 17.8 months (range: 0.2 - 39.1 months) there were 289 PFS events for Welireg and 276 PFS events for everolimus. The median PFS was 5.6 months (95% CI: 3.8, 6.5) for Welireg versus 5.6 months (95% CI: 4.8, 5.8) for everolimus. The PFS hazard ratio was 0.74 (95% CI: 0.63, 0.88) (Figure 2). The median duration of response was 19.5 (range: 1.9 - 31.6+) for Welireg versus 13.7 (range: 3.8 - 21.2+) for everolimus. Based on Kaplan-Meier estimates, patients with a DOR ≥ 12 months was 73% for Welireg versus 62% for everolimus. At the pre-specified interim analysis, OS favoured belzutifan over everolimus, but did not reach statistical significance.
At a subsequent pre-specified analysis with median follow-up time of 17.8 months (range: 0.2 - 39.1 months) there were 289 PFS events for Welireg and 276 PFS events for everolimus. The median PFS was 5.6 months (95% CI: 3.8, 6.5) for Welireg versus 5.6 months (95% CI: 4.8, 5.8) for everolimus. The PFS hazard ratio was 0.74 (95% CI: 0.63, 0.88) (Figure 2). The median duration of response was 19.5 (range: 1.9 - 31.6+) for Welireg versus 13.7 (range: 3.8 - 21.2+) for everolimus. Based on Kaplan-Meier estimates, patients with a DOR ≥ 12 months was 73% for Welireg versus 62% for everolimus. At the pre-specified interim analysis, OS favoured belzutifan over everolimus, but did not reach statistical significance.
The median Time to Response (TTR) was 3.8 months (range: 1.7 - 22.0) in the belzutifan group and 3.7 months (range: 1.8 - 5.4) in the everolimus group. ORR analysis demonstrated ORR of 22.7% for Welireg versus 3.5% for everolimus.
5.2 Pharmacokinetic Properties
The pharmacokinetics of Welireg are similar in healthy subjects and patients with solid tumours including advanced RCC. Cmax and AUC increase proportionally over a dose range of 20 mg to 120 mg. Based on population-PK analysis, the simulated geometric mean steady-state (CV%) Cmax is 1.5 microgram/mL (46%) and AUC0-24hr is 20.8 microgram.hr/mL (64%) in patients treated with 120 mg belzutifan. Steady-state is reached after approximately 3 days.
Absorption.
Following single-dose oral administration of 120 mg of Welireg, peak plasma concentrations (median Tmax) of belzutifan occurred at 1 to 2 hours postdose.
Effect of food.
A high-fat, high-calorie meal delayed peak belzutifan concentration by approximately 2 hours but, had no effect on exposure (AUC). There was a modest decrease of Cmax by 35% following consumption of a high-fat, high-calorie meal, but this was not clinically meaningful. Therefore, Welireg can be taken without regard to food.
Distribution.
Based on the population PK analysis, the mean (CV%) volume of distribution is 120 L (28.5%). Plasma protein binding of belzutifan is 45%. The blood-to-plasma concentration ratio of belzutifan is 0.88.
Metabolism.
Belzutifan is primarily metabolised by UGT2B17 and CYP2C19 and to a lesser extent by CYP3A4. Both UGT2B17 and CYP2C19 display genetic polymorphisms (see Section 5.1 Pharmacodynamic Properties).
Excretion.
Based on the population PK analysis, the mean (CV%) clearance is 5.89 L/hr (60.6%) and the mean elimination half-life is approximately 14 hrs.
Following oral administration of radiolabelled belzutifan to healthy subjects, approximately 49.6% of the dose was excreted in urine and 51.7% in feces (primarily as inactive metabolites). Approximately 6% of the dose was recovered as parent drug in urine.
Special populations.
Renal impairment.
No relevant increase in exposure (AUC) was observed for subjects with mild or moderate renal impairment. Renal impairment (as evaluated by eGFR) was not identified as a significant covariate in the population pharmacokinetic analysis. The pharmacokinetics of belzutifan have not been studied in patients with severe renal impairment (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Hepatic impairment.
No relevant increase in exposure (AUC) was observed for subjects with mild hepatic impairment (using NCI index) based on population pharmacokinetic analysis. The pharmacokinetics of belzutifan have not been studied in patients with moderate or severe hepatic impairment (see Section 4.2 Dose and Method of Administration; Section 4.4 Special Warnings and Precautions for Use).
Dual UGT2BI7 and CYP2C19 poor metabolisers.
Patients who are poor metabolisers of UGT2B17 and CYP2C19 had higher belzutifan AUC (see Section 5.1 Pharmacodynamic Properties).
Paediatric.
No studies with Welireg have been performed in pediatric patients.
Effects of age, gender, ethnicity, race, and bodyweight. Based on a population pharmacokinetic analysis, age, gender, ethnicity, race, and bodyweight do not have a clinically meaningful effect on the pharmacokinetics of belzutifan. Potential differences in exposure across races are possible due to different frequencies of metabolising enzymes (see Section 5.1 Pharmacodynamic Properties).
Drug interaction studies. In vitro assessment of drug interactions.
Belzutifan is a substrate of UGT2B17, CYP2C19 and CYP3A4. Active transport is not an important determinant of belzutifan disposition. Belzutifan is not an inhibitor of CYP enzymes, UGT enzymes or transporters with the exception of MATE2K. Belzutifan does not induce CYP1A2 or CYP2B6, however, belzutifan induces CYP3A4 in a concentration dependent manner.
In vivo assessment of drug interactions.
In a clinical study, repeat administration of Welireg 120 mg QD resulted in a 40% reduction in midazolam AUC, an effect consistent with a weak CYP3A4 inducer. Based on PBPK modeling, Welireg may exhibit moderate CYP3A4 induction in patients who have higher belzutifan plasma exposures (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions; Section 5.1 Pharmacodynamic Properties).
5.3 Preclinical Safety Data
Genotoxicity.
Belzutifan was not genotoxic in in vitro bacterial mutagenicity assay and micronucleus assay in human lymphocytes, and an in vivo rat micronucleus assay.
Carcinogenicity.
Carcinogenicity studies have not been conducted with belzutifan.6 Pharmaceutical Particulars
6.1 List of Excipients
Welireg tablets contain the inactive ingredients: croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, mannitol, microcrystalline cellulose, and silicon dioxide.
The film-coat contains indigo carmine aluminium lake, macrogol, polyvinyl alcohol, purified talc, titanium dioxide.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C.
6.5 Nature and Contents of Container
Welireg is available in the following presentations:
In HDPE bottle with child-resistant closure in pack size of 90 tablets.
In PVC coated aluminium foil blisters in pack size of 90 (3 packs of 30) tablets.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Belzutifan is a white to light brown powder that is soluble in acetonitrile, dimethoxyethane and acetone, sparingly soluble in ethyl acetate, very slightly soluble in isopropanol and toluene, and insoluble in water.
Chemical structure.
The chemical name of belzutifan is 3-[[(1S,2S,3R)-2,3-Difluoro-2,3-dihydro-1-hydroxy-7- (methylsulfonyl)-1H-inden-4-yl]oxy]-5-fluorobenzonitrile. The molecular formula is C17H12F3NO4S and the molecular weight is 383.34 Daltons.
The chemical structure is:

CAS number.
1672668-24-4.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (Schedule 4).
Summary Table of Changes
