

What is in this leaflet
This leaflet answers some common questions about XALKORI.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking XALKORI against the benefits it is expected to have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What XALKORI is used for
XALKORI is used to treat rare types of lung cancer caused by defects in a gene called anaplastic lymphoma kinase (ALK) or a gene called ROS1.
XALKORI may slow or stop the growth of these types of lung cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
XALKORI is only available with a doctor's prescription.
It is not addictive.
Use in Children
The safety and efficacy of XALKORI in children have not been established.
Before you take XALKORI
When you must not take it
Do not take XALKORI if you have an allergy to:
- crizotinib (the active ingredient in XALKORI)
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take XALKORI after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have ever had any of the following medical conditions:
- any other lung problems
- problems with your heart rate or heart rhythm, including prolonged QT interval
- liver problems
- kidney problems
- inflammation of the gastrointestinal tract
- cancer that has spread to the stomach or intestines
Tell your doctor if you are pregnant, plan to become pregnant or plan to father a child. XALKORI may affect the developing baby.
Tell your doctor if you are breast-feeding. You should not breastfeed if you are being treated with XALKORI.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including:
- all prescription medicines
- all medicines, vitamins, herbal supplements or natural therapies you buy without a prescription from a pharmacy, supermarket, naturopath or health food shop.
Some medicines may be affected by XALKORI or may affect how well it works.
Tell your doctor or pharmacist if you are taking any of the following medicines:
- itraconazole, ketoconazole or voriconazole, medicines to treat fungal infections
- atazanavir, efavirenz, indinavir, ritonavir or saquinavir, medicines to treat HIV infection/AIDS
- clarithromycin, troleandomycin, moxifloxacin or ciprofloxacin, medicines to treat bacterial infections
- carbamazepine, phenobarbital or phenytoin, medicines to treat epilepsy, seizures or fits
- rifabutin or rifampicin, medicines to treat tuberculosis and some other infections
- St. John's wort, a herbal medicine to treat anxiety or depression
- ergotamine, a medicine to treat migraine and some other types of headache
- chlorpromazine, amisulpride, droperidol, haloperidol or ziprasidone, medicines to treat mental illness
- amitriptyline, citalopram, escitalopram, fluoxetine, imipramine or other medicines to treat depression
- midazolam, a medicine used for sedation or to cause sleepiness
- alfentanil, fentanyl or methadone, medicines to treat pain
- ciclosporin, sirolimus or tacrolimus, medicines used in transplant patients
- amiodarone, digoxin, diltiazem, disopyramide, verapamil, bisoprolol, carvedilol or sotalol, medicines to treat heart problems and/or high blood pressure
- clonidine, atenolol, labetalol, metoprolol, nebivolol, oxprenolol, pindolol or propranolol, medicines to treat high blood pressure and/or other heart problems
- colchicine, a medicine to treat gout
- dabigatran, a medicine to prevent blood clots
- bupropion, a medicine to help people stop smoking
- pravastatin, a medicine to treat high cholesterol
- metformin, a medicine to treat diabetes
- mefloquine or quinine, medicines to prevent or treat malaria
You may need to take different amounts of your medicines or you may need to use different medicines. Your doctor will advise you.
How to take XALKORI
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how many capsules you need to take each day. This may depend on your condition and whether you are taking any other medicines.
The usual dose of XALKORI is one capsule of 250 mg taken twice a day.
Your doctor may change your dose during treatment.
How to take it
Swallow the capsule whole with a glass of water.
When to take it
Take the capsules at about the same time each day.
If your doctor has told you to take the capsules twice a day, take one capsule in the morning and one capsule in the evening, about 12 hours apart.
Taking them at the same time each day will help you to remember when to take them.
It does not matter if you take XALKORI before or after food.
If you vomit after taking a dose of XALKORI, do not take an extra dose, just take your next dose at the usual time.
How long to take it
Continue taking XALKORI for as long as your doctor tells you.
If you forget to take it
If it is less than 6 hours before your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking XALKORI as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (Phone 13 11 26) for advice or go to Accident and Emergency at the nearest hospital, if you think you or anyone else may have taken too much XALKORI.
Do this even if there are no signs of discomfort or poisoning.
While you are using XALKORI
Things you must do
Make sure you follow your doctor's instructions and keep all appointments. Your doctor will check your progress. You will also have blood tests and other tests to check for side effects.
Tell your doctor immediately if you or your partner become pregnant while taking XALKORI.
Use contraception (birth control) to prevent pregnancy while you are being treated with XALKORI. Women who could become pregnant or men who could father a child must use a reliable method of contraception during treatment with XALKORI and for at least 90 days after taking the last dose.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
Tell any doctor, dentist or pharmacist who treats you that you are taking XALKORI.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking XALKORI.
Things you must not do
Do not take XALKORI to treat any other complaints unless your doctor tells you to.
Do not give XALKORI to anyone else, even if their condition seems similar to yours.
Do not stop taking XALKORI or change the dosage without checking with your doctor.
Things to be careful of
Avoid eating grapefruit or drinking grapefruit juice while you are being treated with XALKORI. Grapefruit and grapefruit juice may change the amount of XALKORI in your body.
Be careful driving or operating machinery until you know how XALKORI affects you. XALKORI may cause dizziness and tiredness and changes in vision in some people. If you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking XALKORI.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
It can be difficult to tell whether side effects are the result of taking XALKORI, effects of your condition or side effects of other medicines you may be taking. For this reason it is important to tell your doctor of any change in your condition.
Medicines can affect people in different ways.
Ask your doctor or pharmacist to answer any questions you may have.
Do not be alarmed by the list of possible side effects. You may not experience any of them.
Tell your doctor or pharmacist if you notice any of the following:
- blurred vision, flashes of light, floaters in eye, double vision
- nausea (feeling sick) or vomiting
- diarrhoea, constipation
- swelling of hands, feet or legs
- tiredness
- loss of appetite
- dizziness
- tingling or numbness of hands or feet; pins and needles
- change in sense of taste
- soreness or redness of mouth, lips or tongue
- skin rash
- slow heart rate
- pain in back, side or upper stomach.
- heartburn, belching, indigestion
- reduced sex drive in men, difficulty in getting an erection
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital, if you notice any of the following:
- difficulty breathing, shortness of breath, cough, coughing up pinkish mucus or blood, rapid weight gain, fluid retention, swollen ankles
- infection, fever
- dizziness, fainting, chest pain, rapid or irregular heartbeat
- pain on right side of stomach, feeling more tired than usual, weakness, dark urine, itchy skin, yellowing of the skin or eyes, bleeding or bruising more easily than usual
- severe stomach pain
- loss of vision
The above side effects may be serious. You may need urgent medical attention.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some patients.
Some side effects might only be found when your doctor does blood tests or other tests. These tests could show a change in the liver, kidney, levels of blood cells or changes in heart rhythm.
After using XALKORI
Storage
Keep XALKORI capsules in the original container until it is time to take them.
Store XALKORI in a cool dry place where the temperature stays below 30°C.
Do not leave XALKORI or any other medicine in the car or on window sills.
Do not store XALKORI or any other medicine in the bathroom or near a sink. Heat and dampness can destroy some medicines.
Keep the capsules where children cannot reach them. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any capsules that are left over.
Product description
What it looks like
XALKORI 200 mg capsules have a pink cap and white body and are printed with "Pfizer" on the cap and "CRZ 200" on the body in black ink.
XALKORI 250 mg capsules have a pink cap and body and are printed with "Pfizer" on the cap and "CRZ 250" on the body in black ink.
XALKORI is supplied in blister packs containing 60 capsules.
Ingredients
XALKORI capsules contain 200 mg or 250 mg of crizotinib as the active ingredient.
The capsules also contain:
- microcrystalline cellulose
- calcium hydrogen phosphate
- sodium starch glycollate
- magnesium stearate
- colloidal anhydrous silica
- gelatin
- titanium dioxide (E171)
- red iron oxide (E172)
XALKORI capsules do not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Supplier
XALKORI is supplied in Australia by:
Pfizer Australia Pty Ltd
Sydney NSW
Toll Free number: 1800 675 229
Australian registration numbers
XALKORI 200 mg:
AUST R 190964 (blister pack)
XALKORI 250 mg:
AUST R 190965 (blister pack)
Date of preparation
This leaflet was prepared in April 2019.
® = Registered Trademark
© Pfizer Australia Pty Ltd
Published by MIMS June 2019







 Based on IRR assessment, a total of 9 (23.1%) of the 39 patients in the crizotinib arm and 12 (30.0%) of the 40 patients in the chemotherapy arm with previously treated baseline brain metastases experienced progression of intracranial lesions or developed new intracranial lesions. For patients with previously treated baseline brain metastases, the median intracranial TTP (IC-TTP) was 15.7 months in the crizotinib arm and 12.5 months in the chemotherapy arm (HR = 0.45 [95% CI: 0.19, 1.07]; 1 sided p-value = 0.0315). A total of 16 (12.1%) of the 132 patients in the crizotinib arm and 14 (10.7%) of the 131 patients in the chemotherapy arm without baseline brain metastases developed new intracranial lesions. For patients without baseline brain metastases, the median IC-TTP was not reached in either the crizotinib or the chemotherapy arms (HR = 0.69 [95% CI: 0.33, 1.45]; 1 sided p-value = 0.1617).
Based on IRR assessment, a total of 9 (23.1%) of the 39 patients in the crizotinib arm and 12 (30.0%) of the 40 patients in the chemotherapy arm with previously treated baseline brain metastases experienced progression of intracranial lesions or developed new intracranial lesions. For patients with previously treated baseline brain metastases, the median intracranial TTP (IC-TTP) was 15.7 months in the crizotinib arm and 12.5 months in the chemotherapy arm (HR = 0.45 [95% CI: 0.19, 1.07]; 1 sided p-value = 0.0315). A total of 16 (12.1%) of the 132 patients in the crizotinib arm and 14 (10.7%) of the 131 patients in the chemotherapy arm without baseline brain metastases developed new intracranial lesions. For patients without baseline brain metastases, the median IC-TTP was not reached in either the crizotinib or the chemotherapy arms (HR = 0.69 [95% CI: 0.33, 1.45]; 1 sided p-value = 0.1617). The change from baseline scores was found to be significantly different between the 2 treatment arms, with a significantly greater improvement observed in global quality of life in the crizotinib arm compared to the chemotherapy arm (overall difference in change from baseline scores 13.8; p-value < 0.0001).
The change from baseline scores was found to be significantly different between the 2 treatment arms, with a significantly greater improvement observed in global quality of life in the crizotinib arm compared to the chemotherapy arm (overall difference in change from baseline scores 13.8; p-value < 0.0001).
 Patient reported symptoms and global QOL were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13) at baseline (day 1 cycle 1) and day 1 of each subsequent treatment cycle. A total of 162 patients in the crizotinib arm and 151 patients in the chemotherapy arm had completed the EORTC QLQ-C30 and LC13 questionnaires at baseline and at least 1 postbaseline visit.
Patient reported symptoms and global QOL were collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13) at baseline (day 1 cycle 1) and day 1 of each subsequent treatment cycle. A total of 162 patients in the crizotinib arm and 151 patients in the chemotherapy arm had completed the EORTC QLQ-C30 and LC13 questionnaires at baseline and at least 1 postbaseline visit.


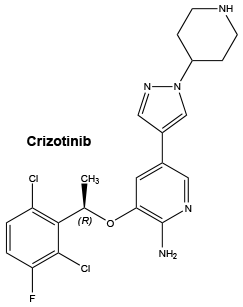 Chemical Name: (R)-3-[1- (2,6-Dichloro-3-fluorophenyl) ethoxy]- 5-[1-(piperidin-4-yl)- 1H-pyrazol-4-yl] pyridin-2-amine.
Chemical Name: (R)-3-[1- (2,6-Dichloro-3-fluorophenyl) ethoxy]- 5-[1-(piperidin-4-yl)- 1H-pyrazol-4-yl] pyridin-2-amine.