



WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about XARELTO. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking XARELTO against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
WHAT XARELTO IS USED FOR
The active substance is rivaroxaban. It belongs to a group of medicines called anticoagulants. It works by inhibiting the blood clotting protein called Factor Xa, thus reducing the tendency of blood to form clots.
XARELTO has been prescribed to you for one of the following uses:
- Prevention of blood clots in your veins after a hip or knee replacement operation because after an operation you are at an increased risk of getting blood clots
- Prevention of blood clots in your brain (stroke) and/or other blood vessels in your body if you have a form of irregular heart rhythm called non-valvular atrial fibrillation
- Treatment of blood clots in the veins of your legs (deep vein thrombosis, DVT) and clots in your lung (pulmonary embolism, PE) and to prevent blood clots from re-occurring in your legs and/or lungs.
XARELTO 2.5 mg tablets twice daily along with aspirin 100 mg once daily, has been prescribed to you for:
- prevention of major heart related events (stroke, heart attack and death from heart related conditions) if you have poor blood flow in the blood vessels of your heart (coronary artery disease or CAD) and/or arms and legs (peripheral artery disease or PAD).
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed XARELTO for another reason.
XARELTO is a prescription medicine. It should only be used in adults under medical supervision.
BEFORE YOU TAKE XARELTO
When you must not take it
Do not take XARELTO if you have an allergy to:
- any medicine containing rivaroxaban
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take XARELTO:
- if you are bleeding excessively or at an increased risk of bleeding
- if you have liver disease which leads to an increased risk of bleeding
- if you have end stage kidney disease or if you are undergoing dialysis (a procedure used to remove waste products from the blood). Your doctor will know how to determine your kidney function.
- If you are taking medicines for fungal infections e.g. ketoconazole, or itraconazole, voriconazole, or posaconazole, unless they are only applied to the skin
- if you are taking anti-viral medicines for HIV/AIDS e.g. ritonavir
- if you had bleeding in the brain within the last 6 months.
If you are not sure whether you should start using XARELTO, talk to your doctor or pharmacist.
Do not use this medicine if you are pregnant or intend to become pregnant. Women should use a reliable contraceptive while taking XARELTO.
Do not use XARELTO if you are breastfeeding. It is not known whether rivaroxaban passes into human breast milk.
Tell your doctor if you have a mechanical heart valve
- XARELTO may not be suitable for you because it has not been studied in patients with mechanical heart valve.
This medicine should not be used in a child under the age of 18 years.
Do not take this medicine after the expiry date printed on the pack and blister. The expiry date is printed on the carton and on each blister after “EXP” (e.g. 11 18 refers to November 2018). The expiry date refers to the last day of that month. If it has expired, return it to your pharmacist for disposal.
Do not take this medicine if the packaging is torn or shows signs of tampering. If the packaging is damaged, return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- if you have kidney disease.
Doctor will need to take special care in patients with moderate or severe kidney disease. Your doctor will do tests to determine how severe your kidney disease is. - if you have kidney disease and undergoing dialysis (a procedure used to remove waste products from the blood)
- if you have prosthetic heart valves
- if you have liver disease
- if a doctor has told you that you have a severe form of antiphospholipid syndrome (a disease which can cause blood clots)
- if you have an increased risk of bleeding such as:
- bleeding disorders
- very high blood pressure, not controlled by medical treatment
- an active ulcer or a recent ulcer of your stomach or bowel
- a problem with the blood vessels in the back of your eyes
- recent bleeding in your brain
- a recent operation on your brain, spinal column or eye
- abnormalities in blood vessels of your spine or brain
- a lung disease where your bronchi are widened (bronchiectasis), or history of bleeding in the lungs - you have a coronary artery disease with severe symptomatic heart failure
- if you have CAD and/or PAD and had the following:
- a bleed in your brain (stroke) or
- a blood clot in your brain (ischaemic, non-lacunar stroke) in the previous month or
- a blockage of the small arteries in the brain (lacunar stroke) - if you have an active cancer. An active cancer means that in the last 6 months you:
- have been diagnosed with cancer
- had a relapse of cancer
- were being treated for cancer
Your doctor may decide to keep you under closer observation.
In the event of a surgery
Tell your doctor, dentist or pharmacist if you need to have an operation (including dental work) while you are taking XARELTO. It is very important to take XARELTO and any other medications you might be on, before and after the operation exactly at the times you have been told by your doctor.
During any invasive procedure or operation, if it involves a catheter or injection into your spinal column (e.g. for epidural or spinal anaesthesia or pain reduction):
- it is very important to take XARELTO before and after the injection or removal of the catheter exactly at the times you have been told by your doctor
- tell your doctor immediately if you get numbness or weakness of your legs or problems with your bowel or bladder after the end of anaesthesia, because urgent care is necessary
If you have not told your doctor about any of the above, tell them before you start taking XARELTO.
XARELTO contains lactose. If you have been told by your doctor that you have an intolerance to some sugars, contact your doctor before taking it.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Your doctor or pharmacist may have more information on medicines to be careful with or to avoid while taking XARELTO because its effect may be increased.
Some medicines and XARELTO may interfere with each other. These include:
- other medicines to reduce blood clotting e.g. enoxaparin, clopidogrel or warfarin
- some medicines to treat depression (selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs))
These medicines may be affected by XARELTO, may increase the effect of XARELTO or mayaffect how well XARELTO works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking XARELTO.
Tell your doctor if you are taking anti-inflammatory and pain relieving medicines e.g. naproxen or medicines used for the protection of heart disease e.g. acetylsalicylic acid (aspirin).
Your doctor may decide to keep you under closer observation. If your doctor thinks that you are at increased risk of developing stomach or bowel ulcers, he may also use a preventative ulcer treatment.
If you have CAD and/or PAD, your doctor may ask you to take your XARELTO 2.5 mg tablet twice daily with aspirin 100 mg once daily.
If you are taking
- medicines for treatment of epilepsy (phenytoin, carbamazepine)
- St John’s Wort, a herbal product used for depression
- Rifampicin, an antibiotic
Tell your doctor before taking XARELTO, because its effect may be reduced.
HOW TO TAKE XARELTO
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the pharmacist label, ask your doctor or pharmacist for help.
How much to take
To prevent clots in your veins after a hip or knee replacement operation, the dose is one XARELTO 10 mg tablet once a day with or without food.
To prevent blood clots in brain (stroke) and other blood vessels, the usual dose is one XARELTO 20 mg tablet once daily. If your kidneys are not working properly, your doctor may reduce your dose to one XARELTO 15 mg tablet once daily. The tablet packs are marked with days of the week to help you remember if you have taken your daily dose. XARELTO 15 mg and 20 mg tablets are to be taken with food.
To treat blood clots in your legs and clots in your lungs and for preventing blood clots from re-occurring, the usual dose is one XARELTO 15 mg tablet TWICE daily for the first three weeks, followed by one XARELTO 20 mg tablet ONCE daily. The initial treatment pack (42 tablet pack) is marked with days of the week and “am” for the morning dose and “pm” for the evening dose. This will help you remember if you have taken the required dose. After 6 to 12 months treatment your doctor may decide to continue treatment with either one XARELTO 20 mg tablet ONCE a day or one XARELTO 10 mg tablet ONCE a day. XARELTO 15 mg and 20 mg tablets are to be taken with food.
To prevent major heart related events if you have CAD and/or PAD, the dose is one XARELTO 2.5 mg tablet twice daily. You must also take one 100 mg aspirin tablet once a day. XARELTO 2.5 mg tablets can be taken with or without food. Swallow the tablets preferably with water.
If you have difficulty swallowing the tablet whole, talk to your doctor about other ways to take XARELTO. The tablet may be crushed and mixed with water or apple puree immediately before you take it. This drink should be immediately followed by food.
If necessary, the crushed XARELTO tablet may be given to you through a stomach tube.
After giving the crushed XARELTO tablet via the stomach tube, you or your carer should flush the tube with water. If you are taking the 15 or 20 mg XARELTO tablet, you should be fed via the stomach tube straight after your dose of XARELTO.
When to take it
Following hip or knee replacement operation: Take the first tablet 6 to10 hours after your operation or as advised by your doctor. Then take a tablet every day for the duration prescribed, unless your doctor tells you to stop.
If you have had a hip replacement you will usually take the tablets for 5 weeks. If you have had a knee replacement you will usually take the tablets for 2 weeks. Your doctor will advise you about the exact duration.
For prevention of stroke or treatment or prevention of blood clots in your legs and/or lungs, take the tablet(s) every day until your doctor tells you to stop. Your doctor will decide how long you must continue your treatment.
If your heart beat needs to be restored to normal by a procedure called cardioversion, take XARELTO according to your doctor’s instructions.
If you need a procedure to treat blocked blood vessels in your heart (called a percutaneous coronary intervention – PCI with an insertion of a stent), the dose may be changed by your doctor. Your doctor will advise you about any changes to the amount of XARELTO you should take.
For prevention of major heart related events if you have CAD and/or PAD, your doctor will tell you when to start treatment with XARELTO 2.5 mg twice daily with aspirin 100 mg once daily. Your doctor will decide how long you must continue treatment.
It is important that you follow instructions from your doctor and not to miss or stop taking your medicine or lower the dosage without checking with your doctor. XARELTO has been prescribed to you by your doctor to treat and/or prevent a serious medical condition.
Try to take the tablet(s) at the same time every day to help you remember.
If you forget to take it
If you are taking one XARELTO 10 mg, or one 15 mg, or one 20 mg tablet ONCE a day: If you have missed a dose, take it as soon as you remember. Do not take more than one tablet in a single day to make up for a forgotten dose. Take the next tablet on the following day and then carry on taking a tablet once a day as normal. Do not take a double dose to make up for a forgotten tablet.
If you are taking one XARELTO 15 mg tablet TWICE a day and have missed a dose, take it as soon as you remember. If you forget to take a dose; you can take two XARELTO 15 mg tablets at the same time to get a total dose of 30 mg in one day. The following day onwards, you should take one XARELTO 15 mg tablet twice a day as normal, until required.
If you are taking XARELTO 2.5 mg tablet TWICE a day and have missed a dose, you can take the next dose at the usual time. Do not take a double dose to make up for a missed dose.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (Australia: 13 11 26 or New Zealand: 0800 POISON or 0800 764 766) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much XARELTO. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Taking too much XARELTO increases the risk of bleeding.
WHILE YOU ARE TAKING XARELTO
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking XARELTO.
Take XARELTO exactly as your doctor has prescribed.
Remember to carry your patient alert card in your wallet with you at all times.
Tell all doctors, dentists and pharmacists who are treating you that you are taking XARELTO.
Tell your doctor if you need to have a surgical or dental procedure.
Tell your doctor that you are using XARELTO, if your doctor is planning for you to have an anaesthetic injection in your back (spinal or epidural injection).
Tell your doctor if other medications are prescribed to you during the course of therapy with XARELTO.
If you become pregnant while you are taking XARELTO, immediately tell your doctor.
Things you must not do
Do not take XARELTO to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor first because XARELTO treats and prevents serious conditions.
Things to be careful of
If this medicine makes you feel faint or dizzy, do not drive or use machinery.
SIDE EFFECTS
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking XARELTO.
All medicines have side effects. Sometimes they are serious, most of the time they are not. In serious cases, you may need medical attention.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Your doctor may need to monitor and conduct blood tests, as XARELTO can affect your liver or pancreatic enzymes. You may not experience any specific symptoms.
Like other similar medicines (anticoagulants), XARELTO may cause bleeding, which may potentially be life threatening. In some cases this bleeding may not be obvious. There is no antidote available to reverse the effects of XARELTO, however there are measures your health professional can take to control / stop the bleeding. Please see your doctor if you experience any symptoms of bleeding.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- exceptional weakness, unexplained swelling
- breathlessness, chest pain
- signs of allergy such as rash, itching or hives on skin, swelling of the face, lips, tongue, or other parts of the body, shortness of breath, wheezing or trouble breathing
- signs of liver problems such as yellowing of the skin and/or eyes (jaundice)
- prolonged or excessive bleeding from gums, nose etc
- numbness in the arms and legs
- dizziness, fainting
- oozing from a surgical wound
- vomiting or coughing up blood
- blood in the urine or stool
- heavy menstrual bleeding
- skin condition with severe blisters and bleeding in the lips, eyes, mouth, nose and genitals
- extensive skin rash associated with fever
- blood in urine, reduced urine output, swelling of the ankles, feet and legs, increased time for blood to clot, and heavy bleeding. These symptoms may be related to a condition called anticoagulant-related nephropathy.
Your doctor may decide to keep you under observation or change how you should be treated.
Tell your doctor as soon as possible if you experience any of the following side effects and they worry you.
- tiredness, pale skin and breathlessness
- bruising
- feeling sick (nausea)
- headache
- diarrhoea, indigestion, or stomach pain
- pain in the arms or legs
- constipation
- fever
- frequent infections such as severe chills, sore throat and mouth ulcers
- mild rash, itchy skin
- fast heart beat
These side effects are usually mild.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people. If you notice any side effects not listed in this leaflet, please tell your doctor or pharmacist.
AFTER USING XARELTO
Storage
Keep your tablets in their blister pack until it is time to take them. If you take the tablets out of the box or blister pack, they may not keep well.
Store the tablets in a cool dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom, near a sink, or on a window sill.
Do not leave it in the car. Heat and damp can destroy some medicines.
Keep out of the reach and sight of children. A locked cupboard at least one and a half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking the tablets, or the tablets have passed their expiry date, ask your pharmacist what to do with any that are left over.
Return any unused medicine to your pharmacist.
PRODUCT DESCRIPTION
What it looks like
XARELTO 2.5 mg film-coated tablets are light yellow, round, film-coated tablets marked with the BAYER cross on one side and "2.5" and a triangle on the other side. It is packed in blister packs in cartons of 14, 56, 60 and 100 tablets.
XARELTO 10 mg film-coated tablets are light red, round, film-coated tablets marked with the BAYER-cross on one side and “10” and a triangle on the other side. It is packed in blister packs in cartons of 3, 10, 15, 30 and 100 tablets.
XARELTO 15 mg film-coated tablets are red, round, film-coated tablets marked with the BAYER-cross on one side and “15” and a triangle on the other side. It is packed in blister packs in cartons of 7, 14, 28, 42, 84, 98 and 100 tablets.
XARELTO 20 mg film-coated tablets are brown- red, round, film-coated tablets marked with the BAYER-cross on one side and “20” and a triangle on the other side. It is packed in blister packs in cartons of 7, 28, 84, 98 and 100 tablets.
Not all pack sizes may be marketed.
Ingredients
Active ingredient per tablet:
- XARELTO 2.5 mg contains 2.5 mg rivaroxaban
- XARELTO 10 mg contains 10 mg rivaroxaban
- XARELTO 15 mg contains 15 mg rivaroxaban
- XARELTO 20 mg contains 20 mg rivaroxaban
Inactive ingredients:
- microcrystalline cellulose
- croscarmellose sodium
- hypromellose
- lactose monohydrate
- magnesium stearate
- sodium lauryl sulfate
- iron oxide red (10 mg, 15 mg and 20 mg tablets)
- iron oxide yellow (2.5 mg tablets)
- macrogol 3350
- titanium dioxide
Supplier
Made in Germany or Italy for:
Bayer Australia Ltd
ABN 22 000 138 714
875 Pacific Highway
Pymble NSW 2073
Bayer New Zealand Limited
PO Box 2825
Shortland St
Auckland 1140
New Zealand
Telephone: 0800 233 988
Australian Registration Number
XARELTO 2.5 mg - AUST R 298198
XARELTO 10 mg - AUST R 147400
XARELTO 15 mg - AUST R 181185
XARELTO 20 mg - AUST R 181186
Date of Preparation
May 2023
See TGA website (www.ebs.tga.gov.au) for latest Australian Consumer Medicine Information.
See MEDSAFE website (www.medsafe.govt.nz) for latest New Zealand Consumer Medicine Information.
® Registered Trademark of Bayer Group, Germany
© Bayer Australia Ltd All rights reserved.

Published by MIMS July 2023




 See Table 6.
See Table 6. Due to the pharmacological mode of action, the use of Xarelto may be associated with an increased risk of occult or overt bleeding from any tissue and organ which may result in posthaemorrhagic anaemia. The signs, symptoms, and severity (including possible fatal outcome) will vary according to the location and degree or extent of the bleeding and/or anaemia. The risk of bleedings may be increased in certain patient groups, e.g. patients with uncontrolled severe arterial hypertension and/or taking concomitant medications affecting haemostasis (see Section 4.4 Special Warnings and Precautions for Use).
Due to the pharmacological mode of action, the use of Xarelto may be associated with an increased risk of occult or overt bleeding from any tissue and organ which may result in posthaemorrhagic anaemia. The signs, symptoms, and severity (including possible fatal outcome) will vary according to the location and degree or extent of the bleeding and/or anaemia. The risk of bleedings may be increased in certain patient groups, e.g. patients with uncontrolled severe arterial hypertension and/or taking concomitant medications affecting haemostasis (see Section 4.4 Special Warnings and Precautions for Use).


 The activated partial thromboplastin time (aPTT) and HepTest are also prolonged dose dependently; however, they are not recommended to assess the pharmacodynamic effect of rivaroxaban. Anti-factor Xa activity is influenced by rivaroxaban (see Section 4.4 Special Warnings and Precautions for Use).
The activated partial thromboplastin time (aPTT) and HepTest are also prolonged dose dependently; however, they are not recommended to assess the pharmacodynamic effect of rivaroxaban. Anti-factor Xa activity is influenced by rivaroxaban (see Section 4.4 Special Warnings and Precautions for Use). The respective studies were heterogeneous with respect to their composition of participating countries (centres from Europe, North and South America, Asia and Australia). Men and women of 18 years or older scheduled for hip or knee replacement surgery could be enrolled provided that they had no active or high risk of bleeding or other conditions contraindicating treatment with low molecular weight heparin, no significant liver disease, were not pregnant or breastfeeding, or were not using HIV protease inhibitors.
The respective studies were heterogeneous with respect to their composition of participating countries (centres from Europe, North and South America, Asia and Australia). Men and women of 18 years or older scheduled for hip or knee replacement surgery could be enrolled provided that they had no active or high risk of bleeding or other conditions contraindicating treatment with low molecular weight heparin, no significant liver disease, were not pregnant or breastfeeding, or were not using HIV protease inhibitors.
 Exclusion criteria included:
Exclusion criteria included:
 In addition to the phase III ROCKET AF study, a prospective, single-arm, post-authorisation, non-interventional, open-label cohort study (XANTUS) with central outcome adjudication including thromboembolic events and major bleeding has been conducted, wherein 6,785 patients with non-valvular atrial fibrillation were enrolled for prevention of stroke and non-central nervous system (CNS) systemic embolism under real-world conditions (safety analysis set n = 6,784). The mean CHADS2 score was 2.0 compared to a mean CHADS2 score of 3.5 in ROCKET AF. Major bleeding occurred in 2.1 per 100 patient years. Fatal haemorrhage was reported in 0.2 per 100 patient years and intracranial haemorrhage in 0.4 per 100 patient years. Stroke or non-CNS systemic embolism was recorded in 0.8 per 100 patient years. These observations from routine clinical practice are consistent with the results observed in the ROCKET AF study.
In addition to the phase III ROCKET AF study, a prospective, single-arm, post-authorisation, non-interventional, open-label cohort study (XANTUS) with central outcome adjudication including thromboembolic events and major bleeding has been conducted, wherein 6,785 patients with non-valvular atrial fibrillation were enrolled for prevention of stroke and non-central nervous system (CNS) systemic embolism under real-world conditions (safety analysis set n = 6,784). The mean CHADS2 score was 2.0 compared to a mean CHADS2 score of 3.5 in ROCKET AF. Major bleeding occurred in 2.1 per 100 patient years. Fatal haemorrhage was reported in 0.2 per 100 patient years and intracranial haemorrhage in 0.4 per 100 patient years. Stroke or non-CNS systemic embolism was recorded in 0.8 per 100 patient years. These observations from routine clinical practice are consistent with the results observed in the ROCKET AF study. EINSTEIN-DVT (see Table 17) met its principal objective, demonstrating that Xarelto was noninferior to enoxaparin/ VKA for the primary outcome of symptomatic recurrent VTE (HR of 0.68 [95% CI = 0.44-1.04], p < 0.001). The prespecified test for superiority was not statistically significant (p = 0.0764). The incidence rates for the principal safety outcome (major or clinically relevant nonmajor bleeding events), as well as the secondary safety outcome (major bleeding events), were similar for both groups (HR of 0.97 [95% CI = 0.76-1.22], p = 0.77 and HR of 0.65 [95% CI = 0.33-1.30], p = 0.21, respectively). The predefined secondary outcome of net clinical benefit, (the composite of the primary efficacy outcome and major bleeding events), was reported with a HR of 0.67 ([95% CI = 0.47-0.95], p = 0.03) in favour of Xarelto.
EINSTEIN-DVT (see Table 17) met its principal objective, demonstrating that Xarelto was noninferior to enoxaparin/ VKA for the primary outcome of symptomatic recurrent VTE (HR of 0.68 [95% CI = 0.44-1.04], p < 0.001). The prespecified test for superiority was not statistically significant (p = 0.0764). The incidence rates for the principal safety outcome (major or clinically relevant nonmajor bleeding events), as well as the secondary safety outcome (major bleeding events), were similar for both groups (HR of 0.97 [95% CI = 0.76-1.22], p = 0.77 and HR of 0.65 [95% CI = 0.33-1.30], p = 0.21, respectively). The predefined secondary outcome of net clinical benefit, (the composite of the primary efficacy outcome and major bleeding events), was reported with a HR of 0.67 ([95% CI = 0.47-0.95], p = 0.03) in favour of Xarelto. In the EINSTEIN PE study (see Table 18) rivaroxaban was demonstrated to be noninferior to enoxaparin/ VKA for the primary efficacy outcome (p = 0.0026 (test for noninferiority); hazard ratio: 1.12 (0.75-1.68)). The prespecified net clinical benefit (primary efficacy outcome plus major bleeding events) was reported with a hazard ratio of 0.85 ((95% CI: 0.63-1.14), nominal p-value p = 0.275).
In the EINSTEIN PE study (see Table 18) rivaroxaban was demonstrated to be noninferior to enoxaparin/ VKA for the primary efficacy outcome (p = 0.0026 (test for noninferiority); hazard ratio: 1.12 (0.75-1.68)). The prespecified net clinical benefit (primary efficacy outcome plus major bleeding events) was reported with a hazard ratio of 0.85 ((95% CI: 0.63-1.14), nominal p-value p = 0.275).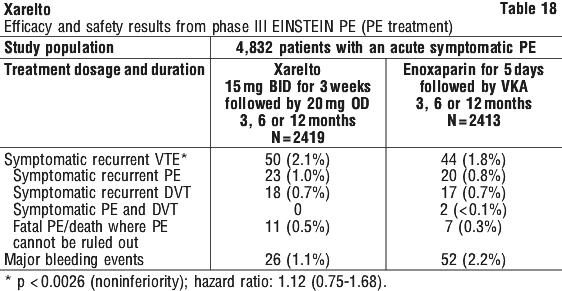 A prespecified pooled analysis of the outcome of the EINSTEIN DVT and PE studies was conducted (see Table 19).
A prespecified pooled analysis of the outcome of the EINSTEIN DVT and PE studies was conducted (see Table 19).
 In terms of other secondary outcomes, vascular events occurred in 3 patients in the Xarelto arm and 4 patients in the placebo group (HR of 0.74 [95% CI = 0.17-3.3], p = 0.69), and total mortality accounted for 1 (0.2%) vs. 2 (0.3%) of patients in the Xarelto vs. placebo arms, respectively.
In terms of other secondary outcomes, vascular events occurred in 3 patients in the Xarelto arm and 4 patients in the placebo group (HR of 0.74 [95% CI = 0.17-3.3], p = 0.69), and total mortality accounted for 1 (0.2%) vs. 2 (0.3%) of patients in the Xarelto vs. placebo arms, respectively. In addition to the phase III EINSTEIN program, a prospective, non-interventional, open-label cohort study (XALIA) with central outcome adjudication including recurrent VTE, major bleeding and death has been conducted. 5,142 patients with acute DVT were enrolled to investigate the long-term safety of rivaroxaban compared with standard-of-care anticoagulation therapy under real-world conditions. In the safety analysis set (n = 4,768), rates of major bleeding, recurrent VTE and all-cause mortality for rivaroxaban were 0.7%, 1.4% and 0.5%, respectively. There were differences in patient baseline characteristics including age, cancer and renal impairment. A pre-specified propensity score stratified analysis was used to adjust for measured baseline differences but residual confounding may, in spite of this, influence the results. Adjusted hazard ratios comparing rivaroxaban and standard-of-care for major bleeding, recurrent VTE and all-cause mortality were 0.77 (95% CI: 0.40-1.50, p = 0.44), 0.91 (95% CI: 0.54-1.54, p = 0.72) and 0.51 (95% CI: 0.24-1.07, p = 0.074), respectively.
In addition to the phase III EINSTEIN program, a prospective, non-interventional, open-label cohort study (XALIA) with central outcome adjudication including recurrent VTE, major bleeding and death has been conducted. 5,142 patients with acute DVT were enrolled to investigate the long-term safety of rivaroxaban compared with standard-of-care anticoagulation therapy under real-world conditions. In the safety analysis set (n = 4,768), rates of major bleeding, recurrent VTE and all-cause mortality for rivaroxaban were 0.7%, 1.4% and 0.5%, respectively. There were differences in patient baseline characteristics including age, cancer and renal impairment. A pre-specified propensity score stratified analysis was used to adjust for measured baseline differences but residual confounding may, in spite of this, influence the results. Adjusted hazard ratios comparing rivaroxaban and standard-of-care for major bleeding, recurrent VTE and all-cause mortality were 0.77 (95% CI: 0.40-1.50, p = 0.44), 0.91 (95% CI: 0.54-1.54, p = 0.72) and 0.51 (95% CI: 0.24-1.07, p = 0.074), respectively.




 Rivaroxaban is an odourless, non-hygroscopic white to yellowish powder. Rivaroxaban is practically insoluble in water and aqueous media in the pH range 1 to 9. An amount of approximately 5-7 mg/L rivaroxaban is pH-independently soluble in aqueous media at 25°C. Rivaroxaban is only slightly soluble in organic solvents (e.g. acetone, polyethylene glycol 400).
Rivaroxaban is an odourless, non-hygroscopic white to yellowish powder. Rivaroxaban is practically insoluble in water and aqueous media in the pH range 1 to 9. An amount of approximately 5-7 mg/L rivaroxaban is pH-independently soluble in aqueous media at 25°C. Rivaroxaban is only slightly soluble in organic solvents (e.g. acetone, polyethylene glycol 400).