
What is in this leaflet
This leaflet answers some common questions about XERMELO. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking XERMELO against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What XERMELO is used for
This medicine contains the active substance telotristat ethyl.
XERMELO is used in adults for the treatment of a condition called 'carcinoid syndrome'. This is when a tumour, called a 'neuroendocrine tumour', releases a hormone called serotonin into your bloodstream.
When the tumour releases too much serotonin into your bloodstream you can get symptoms such as:
- diarrhoea and stomach (abdominal) pain
- flushing of your skin, particularly the face
- low blood pressure
- rash
- weight loss.
The symptoms are not the same for everyone.
XERMELO works by reducing the amount of serotonin made by the tumour. It will reduce your diarrhoea.
Your doctor will prescribe XERMELO:
- when your tumour has spread to different parts of your body
- and if your diarrhoea is not well controlled with injections of other medicines called 'somatostatin analogues' (lanreotide or octreotide). You should keep having injections of these other medicines when taking XERMELO.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
Before you take XERMELO
When you must not take it
Do not take XERMELO if you have an allergy to:
- telotristat ethyl
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you are pregnant. It is not known how XERMELO may affect the baby.
Do not breast-feed if you are taking this medicine. It is unknown if the active ingredient in XERMELO passes into breast milk and there is a possibility that your baby may be affected.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- if you have liver problems. This is because this medicine is not recommended for use in patients with severe liver problems. Your doctor may decide to decrease your daily dose of XERMELO in cases where your liver problems are considered mild or moderate. Your doctor will also monitor your liver.
- if you have end-stage kidney disease or are on dialysis. This is because this medicine has not been tested in patients with end-stage kidney disease requiring dialysis.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Do not take XERMELO if you are pregnant or might become pregnant. It is not known how XERMELO may affect the baby.
- Women should use effective methods of contraception while taking this medicine.
- Do not breast-feed if you are taking XERMELO, as this medicine may be passed on to your baby.
If you are pregnant or breast-feeding, think you might be pregnant or are planning to have a baby, ask your doctor or pharmacist for advice before taking this medicine.
If you have not told your doctor about any of the above, tell them before you start taking XERMELO.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and XERMELO may interfere with each other. Tell your doctor or pharmacist if you are taking, have taken or are planning to take any of the following:
- medicines for diarrhoea. XERMELO and these medicines reduce the number of your bowel movements and taken together, they can cause severe constipation. Your doctor may need to change the dose of your medicines.
- medicines used to treat epilepsy, such as valproic acid, carbamazepine and topiramate.
- medicines used to treat your neuroendocrine tumour, such as sunitinib or everolimus.
- medicines to treat depression, such as sertraline.
- medicine to treat smoking cessation, such as bupropion.
- medicines used to avoid transplant rejection, such as ciclosporin.
- medicines used to lower cholesterol levels, such as simvastatin and atorvastatin.
- oral contraceptives, such as ethinyloestradiol.
- medicines used to treat high blood pressure, such as amlodipine, nifedipine, felodipine, verapamil and diltiazem.
- medicines used to treat some types of cancers, such as irinotecan, capecitabine and flutamide.
- medicines used to reduce the chance of a blood clot forming, such as prasugrel
- octreotide. If you need treatment with octreotide subcutaneous injection, you should have your injection at least 30 minutes after taking XERMELO.
If any of the above apply to you (or you are not sure), talk to your doctor or pharmacist before taking Xermelo. These medicines may be affected by XERMELO or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take XERMELO
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions in this leaflet, ask your doctor or pharmacist for help.
How much to take
The recommended dose is one tablet three times a day. If needed, your doctor may tell you to take a different dose.
If your symptoms do not improve, talk to your doctor.
How to take it
Swallow the tablets with food or a meal.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
Take your medicine with food
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
Continue taking your medicine until you finish the pack.
Stop using this medicine when the redness and itching have gone.
If you forget to take it
If you forget a dose, take your next dose when it is due, skipping the missed dose.
Do not take a double dose to make up for the dose that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice if you think that you or anyone else may have taken too much XERMELO. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Symptoms of an overdose may include feeling or being sick, diarrhoea or stomach pain.
While you are taking XERMELO
Things you must do
You should keep having injections of somatostatin analogues (lanreotide or octreotide) when taking XERMELO.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking XERMELO.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may carry out blood tests before you start taking this medicine and while you are taking it. This is to check that your liver is working normally.
Things you must not do
Do not take XERMELO to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
Be careful driving or operating machinery until you know how XERMELO affects you. This medicine may cause fatigue in some people. If you have this symptom, do not drive, operate machinery or do anything else that could be dangerous.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking XERMELO.
This medicine helps most people with carcinoid syndrome, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Tell your doctor or pharmacist if you notice any of the following:
Very common side effects:
- Stomach (abdominal) pain
- Feeling tired or weak (fatigue)
- Feeling sick (nausea)
Common side effects:
- Wind
- Fever
- Headache
- Constipation
- Swollen stomach
- Decreased appetite
- Swelling (build-up of fluid in the body)
- Depression. You may experience decreased self-esteem, lack of motivation, sadness or low mood
Uncommon side effects:
- Impacted stools (faecaloma), constipation, watery diarrhoea
- Pale skin (anaemia)
- Vomiting, weight loss
- Back pain or stomach pains particularly after eating or a reduction in passing water (urination)
Tell your doctor immediately if you experience any of the following side effects:
- Breathing problems, rapid heartbeat
- Fever
- Incontinence (uncontrollable urination)
- Confusion, dizziness or agitation.
Tell your doctor or pharmacist as soon as possible if you notice any of the following:
- feeling or being sick, abnormally dark urine, yellow skin or eyes, pain in the upper right belly.
These may be signs that your liver is not working properly. This might also be shown by changes in your blood test results, such as an increase of liver enzymes.
These are serious side effects that may require medical attention. Liver disorders are common.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
After taking XERMELO
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack, they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 30°C.
Do not store XERMELO or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
XERMELO tablets are white to off-white, film-coated and oval shaped. Each tablet is approximately 17 mm long by 7.5 mm wide with 'T-E' debossed on one side and '250' debossed on the other. The tablets are packaged in a PVC/PCTFE/PVC/Aluminium blister. The blisters are packaged in a carton of 90 tablets.
Ingredients
Each XERMELO tablet contains 250 mg of telotristat ethyl (free base) equivalent to 327.9 mg telotristat etiprate as the active ingredient. The inactive ingredients of XERMELO tablets include:
- colloidal anhydrous silica
- croscarmellose sodium
- hyprolose
- lactose
- magnesium stearate
- polyvinyl alcohol
- titanium dioxide (E171)
- macrogol 3350
- purified talc (E553b).
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Sponsor
XERMELO is sponsored in Australia by:
Ipsen Pty Ltd
Level 2, Building 4
Brandon Office Park
540 Springvale Road
Glen Waverley Victoria 3150
Telephone: 1800 317 033
This leaflet was prepared in June 2021
Australian Registration Number:
AUST R 291734
Published by MIMS November 2021

 When the full effect of telotristat is observed (during the last 6 weeks of the DBT period) the proportion of responders with at least 30% BM reduction was 51% (23/45) in the 250 mg group versus 22% (10/45) in the placebo group (post-hoc analysis).
When the full effect of telotristat is observed (during the last 6 weeks of the DBT period) the proportion of responders with at least 30% BM reduction was 51% (23/45) in the 250 mg group versus 22% (10/45) in the placebo group (post-hoc analysis). The proportions of patients reporting reductions from baseline in daily BM frequency (averaged over 12 weeks) were:
The proportions of patients reporting reductions from baseline in daily BM frequency (averaged over 12 weeks) were: There was no significant difference between treatment groups for the endpoints of flushing and abdominal pain.
There was no significant difference between treatment groups for the endpoints of flushing and abdominal pain.

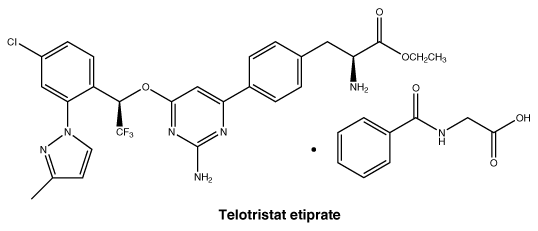 Telotristat etiprate has two asymmetric centres with absolute configuration as indicated in the structure above (R, S configuration).
Telotristat etiprate has two asymmetric centres with absolute configuration as indicated in the structure above (R, S configuration).