What is in this leaflet
This leaflet answers some common questions about XTANDI.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking XTANDI against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What XTANDI is used for
XTANDI contains enzalutamide, an androgen receptor inhibitor, which is used to treat adult men with prostate cancer that:
- no longer responds to hormone therapy or surgical treatment to lower testosterone. Or
- has spread to other parts of the body and is considered "hormone sensitive".
This medicine works by blocking the activity of hormones called androgens (such as testosterone). By blocking androgens, XTANDI stops prostate cancer cells from growing and dividing.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
XTANDI is not for use in children and adolescents.
Safety and effectiveness in children and adolescents have not been established.
Before you take XTANDI
When you must not take it
Do not take XTANDI if you have an allergy to:
- any medicine containing enzalutamide
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
XTANDI is not for use in women.
This medicine may cause harm to the unborn child or potential loss of pregnancy if it is taken by women who are pregnant. It must not be taken by women who are pregnant, may become pregnant, or who are breast feeding.
This medicine could possibly have an effect on male fertility.
If you are having sex with a woman who can become pregnant, you must use a condom and another effective birth control method, during treatment and for 3 months after stopping treatment with this medicine. Men who are sexually active with a pregnant woman must use a condom during and for 3 months after stopping treatment with XTANDI to protect the unborn child.
Female caregivers see section 'How to take Xtandi' for handling and use.
Talk with your doctor if you have questions about birth control. Your doctor can discuss with you the risks and benefits involved.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- a history of seizures
- a serious head injury or a history of head trauma
- a stroke
- a brain tumour, or cancer which has spread to the brain
- drink very large amounts of alcohol either regularly or from time to time
- are taking a medicine that can cause seizures or that increases risks for having seizures (see Taking other medicines)
In some of these situations you may have a higher risk of having a seizure.
Tell your doctor if you have:
- heart or blood pressure problems
- kidney problems
- a partner who is pregnant or is planning to become pregnant.
If you have not told your doctor about any of the above, tell him/ her before you start taking XTANDI.
Taking other medicines
Tell your doctor if you are taking any of the following medicines.
When taken at the same time as XTANDI, these medicines may increase the risk of a seizure:
- certain medicines used to treat asthma and other respiratory diseases (e.g. aminophylline, theophylline)
- medicines used to treat certain psychiatric disorders such as depression and schizophrenia (e.g. clozapine, olanzapine, risperidone, ziprasidone, bupropion, lithium, chlorpromazine, thioridazine, amitriptyline, desipramine, doxepin, imipramine, mirtazapine)
- certain medicines for the treatment of pain (e.g. pethidine)
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and XTANDI may interfere with each other. These include certain medicines used to:
- treat pain (e.g. fentanyl, tramadol, paracetamol)
- thin the blood, or to prevent blood clots (e.g. warfarin, clopidogrel)
- lower cholesterol (e.g. gemfibrozil, atorvastatin, simvastatin)
- treat cancer (e.g. cabazitaxel)
- treat epilepsy (e.g. carbamazepine, clonazepam, phenytoin, primidone, valproic acid)
- treat certain psychiatric disorders such as severe anxiety or schizophrenia (e.g. diazepam, midazolam, haloperidol)
- treat sleep disorders (eg. Zolpidem)
- treat heart conditions or lower blood pressure (e.g. bisoprolol, digoxin, diltiazem, felodipine, nicardipine, nifedipine, propanolol, verapamil)
- treat serious disease related to inflammation (e.g. dexamethasone, prednisolone)
- lower your immunity (e.g. cyclosporin, tacrolimus)
- treat HIV infection (e.g. indinavir, ritonavir)
- treat bacterial infections (e.g. clarithromycin, doxycycline, rifampicin)
- treat thyroid disorders (e.g. levothyroxine)
- treat gout (e.g. colchicine)
- prevent heart conditions or strokes (dabigatran etexilate).
- treat reflux disease or peptic ulcers (e.g. omeprazole)
These medicines may be affected by XTANDI or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take XTANDI
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
The usual dose is four 40 mg capsules taken at the same time once a day. Your doctor may reduce your dose depending on your medical conditions.
Reduced dose:
If you are taking a reduced dose of XTANDI, you may use the remaining capsules in the open dose compartment for your next scheduled dose, provided that the capsules have been otherwise stored under the conditions described below (see After Taking XTANDI, Storage).
How to take it
Swallow the capsules whole with a full glass of water. Do not chew, dissolve or open the capsules before swallowing.
You can take XTANDI with or without food.
XTANDI should not be handled by persons other than the patient and his caregivers. Women who are or may become pregnant should not handle damaged or opened enzalutamide capsules without protection, e.g., gloves.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
It is important to keep taking your medicine even if you feel well.
Do not stop treatment with XTANDI unless your doctor tells you to.
If you forget to take it
Take it as soon as you remember, and then go back to taking your medicine as you would normally.
If you forget to take XTANDI for the whole day, take your usual dose the following day.
If you forget to take XTANDI for more than one day, talk to your doctor immediately.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much XTANDI. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
You may be at increased risk of experiencing a seizure.
While you are taking XTANDI
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking XTANDI.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
If your partner becomes pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked.
Things you must not do
Do not take XTANDI to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or alter the dosage without checking with your doctor.
Do not drive or operate machinery until you know how XTANDI affects you. XTANDI may have a moderate influence on your ability to drive or use any tools or machinery. Seizures have been reported in patients taking XTANDI. If you are at higher risk of seizures, talk to your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking XTANDI.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Seizures
Seizures were reported in 5 in every 1,000 people taking XTANDI and in fewer than one in every 1,000 people taking placebo.
Seizures are more likely if you take more than the recommended dose of this medicine, if you take certain other medicines, or if you are at higher risk of seizure.
If you have a seizure, see your doctor as soon as possible or go to Accident and Emergency at your nearest hospital immediately. Your doctor may decide that you should stop taking XTANDI.
Posterior Reversible Encephalopathy Syndrome (PRES)
There have been rare reports of PRES (may affect up to 1 in 1,000 people), a rare, reversible condition involving the brain, in patients treated with XTANDI. If you have a seizure, worsening headache, confusion, blindness or other vision problems, please contact your doctor as soon as possible.
Tell your doctor or pharmacist if you notice or experience any of the following:
- headache
- weakness
- tiredness
- dizziness
- breathlessness
- chest pain
- swelling of the hands, ankles or feet
- rash
- pain in back, muscles or joints
- hot flushes
- falls
- broken bones
- hallucinations
- feeling anxious
- dry skin
- itching
- high blood pressure
- constipation
- diarrhoea
- feeling sick
- decreased appetite
- difficulty remembering things
- difficulty thinking clearly
- forgetfulness
- reduced concentration.
- cancer as a result of previous treatment with radiation or chemotherapy
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
The above list includes the more common side effects of your medicine.
Other side effects not listed above may also occur in some people.
After taking XTANDI
Storage
Keep your capsules in the pack until it is time to take them. If you take the capsules out of the pack they may not keep as well.
If you are taking a reduced dose of XTANDI, you may store the remaining capsules in the open dose compartment until the next dose, provided that the capsules have been otherwise stored under the conditions described below.
Keep your capsules in the original packaging in a cool dry place where the temperature stays below 25°C.
Do not store XTANDI or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Do not take any capsule that is leaking, damaged, or shows signs of tampering.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Do not throw away any medicines via wastewater or household waste.
Product description
What it looks like
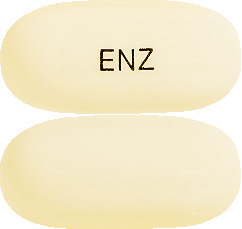
XTANDI capsules are white to off-white, oblong soft gelatin capsules with "ENZ" written on one side in black ink.
XTANDI is available in packs of 112 capsules (in 4 wallets of 28 capsules each).
Ingredients
XTANDI contains 40 mg of enzalutamide as the active ingredient.
Each capsule also contains:
- caprylocaproyl macrogolglycerides
- butylated hydroxyanisole
- butylated hydroxytoluene
- gelatin
- sorbitol sorbitan solution
- glycerol
- titanium dioxide
- purified water
- OPACODE WB monogramming ink NSP-78-17827 BLACK
Supplier
XTANDI is distributed in Australia by:
Astellas Pharma Australia Pty Ltd
6 Eden Park Drive
Macquarie Park, NSW 2113
Medical Information: 1800 751 755
® = Registered Trademark
Australian registration number: AUST R 210494
This leaflet was prepared in March 2021.
Published by MIMS May 2021





 Overall, deaths from adverse reactions during the total duration of treatment occurred in 6 patients (1.7%) receiving Xtandi plus a LHRH analogue, 8 patients (2.3%) receiving Xtandi as monotherapy, and 3 patients (0.8%) receiving placebo plus a LHRH analogue. The reason for death in ≥ 2 patients receiving Xtandi plus a LHRH analogue was infection (n = 2), and the reason for death in ≥ 2 patients receiving Xtandi as monotherapy was arterial thromboembolism (n = 2). Patients receiving placebo plus a LHRH analogue died from sepsis (n = 1), acute myeloid leukemia (n = 1), and myocardial ischaemia (n = 1).
Overall, deaths from adverse reactions during the total duration of treatment occurred in 6 patients (1.7%) receiving Xtandi plus a LHRH analogue, 8 patients (2.3%) receiving Xtandi as monotherapy, and 3 patients (0.8%) receiving placebo plus a LHRH analogue. The reason for death in ≥ 2 patients receiving Xtandi plus a LHRH analogue was infection (n = 2), and the reason for death in ≥ 2 patients receiving Xtandi as monotherapy was arterial thromboembolism (n = 2). Patients receiving placebo plus a LHRH analogue died from sepsis (n = 1), acute myeloid leukemia (n = 1), and myocardial ischaemia (n = 1).





 Xtandi as monotherapy also demonstrated a statistically significant 37% reduction in the risk of developing an MFS event as compared to placebo plus ADT [HR = 0.63 (95% CI: 0.46, 0.87) p = 0.0049]. The median time to an MFS event was not reached in either treatment arm).
Xtandi as monotherapy also demonstrated a statistically significant 37% reduction in the risk of developing an MFS event as compared to placebo plus ADT [HR = 0.63 (95% CI: 0.46, 0.87) p = 0.0049]. The median time to an MFS event was not reached in either treatment arm).

 The major efficacy outcome was supported by multiple secondary endpoints. A statistically significant 72% reduction in risk of initiation of a new antineoplastic therapy was demonstrated for patients in the Xtandi arm compared to patients in the placebo arm (HR = 0.28 [95% CI: 0.20, 0.40]; p < 0.0001). A statistically significant improvement of 19.3% in objective response rate (percentage of patients with complete or partial response in their soft tissue disease) was demonstrated with an Objective Response Rate (OSS) (calculated as percentage of patients with measurable disease at baseline who achieved a complete or partial response in their soft tissue disease) of 83.1% for patients in the Xtandi arm compared to 63.7% for patients in the placebo arm (95% CI: 10.4, 28.2; p < 0.0001). Time to First Symptomatic Skeletal Event (SSE) (time from randomisation to the occurrence of the first symptomatic skeletal event) was associated with a 48% reduction in the risk of a patient experiencing an SSE compared with patients in the placebo arm [HR = 0.52, 95% CI: 0.33, 0.80; nominal p = 0.0026].
The major efficacy outcome was supported by multiple secondary endpoints. A statistically significant 72% reduction in risk of initiation of a new antineoplastic therapy was demonstrated for patients in the Xtandi arm compared to patients in the placebo arm (HR = 0.28 [95% CI: 0.20, 0.40]; p < 0.0001). A statistically significant improvement of 19.3% in objective response rate (percentage of patients with complete or partial response in their soft tissue disease) was demonstrated with an Objective Response Rate (OSS) (calculated as percentage of patients with measurable disease at baseline who achieved a complete or partial response in their soft tissue disease) of 83.1% for patients in the Xtandi arm compared to 63.7% for patients in the placebo arm (95% CI: 10.4, 28.2; p < 0.0001). Time to First Symptomatic Skeletal Event (SSE) (time from randomisation to the occurrence of the first symptomatic skeletal event) was associated with a 48% reduction in the risk of a patient experiencing an SSE compared with patients in the placebo arm [HR = 0.52, 95% CI: 0.33, 0.80; nominal p = 0.0026].

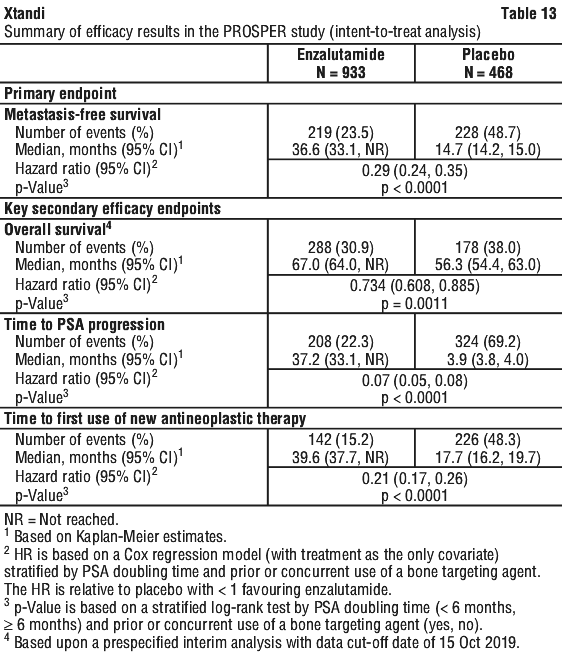
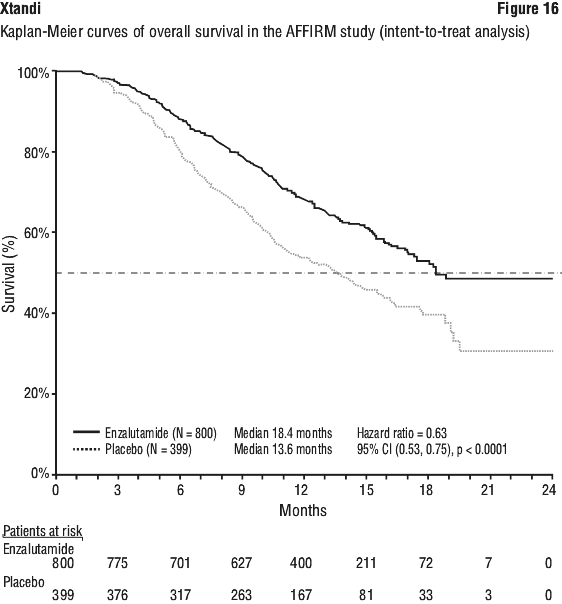
 At the final analysis for overall survival, conducted when 466 deaths were observed, a statistically significant improvement in overall survival was demonstrated in patients randomised to receive enzalutamide compared with patients randomised to receive placebo with a 26.6% reduction in risk of death [hazard ratio (HR) = 0.734, (95% CI: 0.608; 0.885), p = 0.0011]. The median follow-up time was 48.6 and 47.2 months for the enzalutamide and placebo groups, respectively. Thirty-three percent of enzalutamide-treated and 65% of placebo-treated patients received at least one subsequent antineoplastic therapy that may prolong overall survival. See Figure 9.
At the final analysis for overall survival, conducted when 466 deaths were observed, a statistically significant improvement in overall survival was demonstrated in patients randomised to receive enzalutamide compared with patients randomised to receive placebo with a 26.6% reduction in risk of death [hazard ratio (HR) = 0.734, (95% CI: 0.608; 0.885), p = 0.0011]. The median follow-up time was 48.6 and 47.2 months for the enzalutamide and placebo groups, respectively. Thirty-three percent of enzalutamide-treated and 65% of placebo-treated patients received at least one subsequent antineoplastic therapy that may prolong overall survival. See Figure 9. Enzalutamide demonstrated a statistically significant 93% reduction in the relative risk of PSA progression compared to placebo [HR = 0.07 (95% CI: 0.05, 0.08), p < 0.0001]. Median time to PSA progression was 37.2 months (95% CI: 33.1, NR) on the enzalutamide arm versus 3.9 months (95% CI: 3.8, 4.0) on the placebo arm.
Enzalutamide demonstrated a statistically significant 93% reduction in the relative risk of PSA progression compared to placebo [HR = 0.07 (95% CI: 0.05, 0.08), p < 0.0001]. Median time to PSA progression was 37.2 months (95% CI: 33.1, NR) on the enzalutamide arm versus 3.9 months (95% CI: 3.8, 4.0) on the placebo arm.



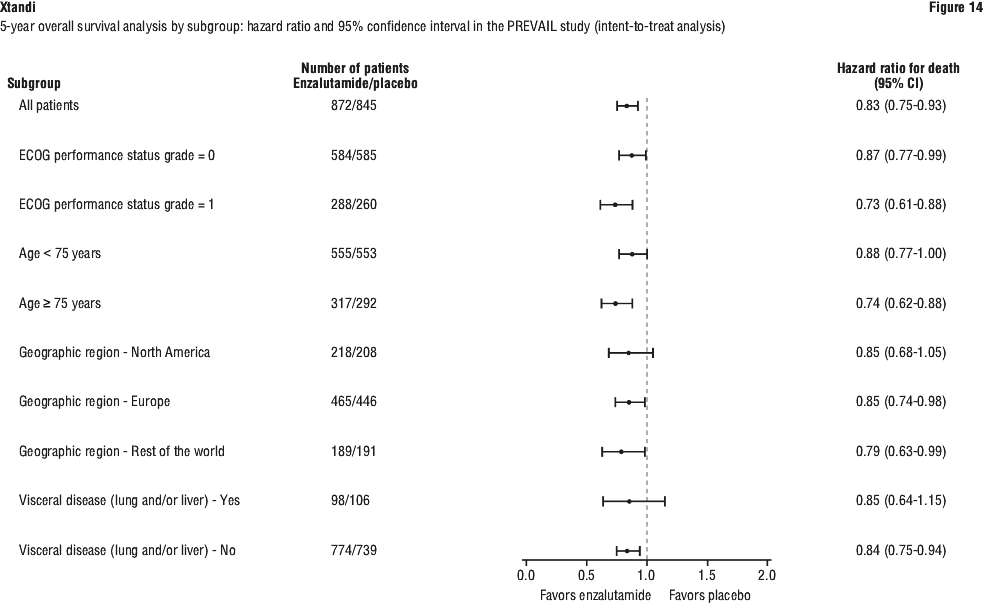 At the pre-specified rPFS analysis, a statistically significant improvement was demonstrated between the treatment groups with an 81.4% reduction in risk of radiographic progression or death [HR = 0.19 (95% CI: 0.15, 0.23), p < 0.0001]. One hundred and eighteen (14%) enzalutamide-treated patients and 321 (40%) of placebo-treated patients had an event. The median rPFS was not reached (95% CI: 13.8, not reached) in the enzalutamide-treated group and was 3.9 months (95% CI: 3.7, 5.4) in the placebo-treated group (see Figure 15). Consistent rPFS benefit was observed across all pre-specified patient subgroups (e.g. age, baseline ECOG performance, baseline PSA and LDH, Gleason score at diagnosis, and visceral disease at screening). A pre-specified follow-up rPFS analysis based on the investigator assessment of radiographic progression demonstrated a statistically significant improvement between the treatment groups with a 69.3% reduction in risk of radiographic progression or death [HR = 0.31 (95% CI: 0.27, 0.35), p < 0.0001]. The median rPFS was 19.7 months in the enzalutamide group and 5.4 months in the placebo group.
At the pre-specified rPFS analysis, a statistically significant improvement was demonstrated between the treatment groups with an 81.4% reduction in risk of radiographic progression or death [HR = 0.19 (95% CI: 0.15, 0.23), p < 0.0001]. One hundred and eighteen (14%) enzalutamide-treated patients and 321 (40%) of placebo-treated patients had an event. The median rPFS was not reached (95% CI: 13.8, not reached) in the enzalutamide-treated group and was 3.9 months (95% CI: 3.7, 5.4) in the placebo-treated group (see Figure 15). Consistent rPFS benefit was observed across all pre-specified patient subgroups (e.g. age, baseline ECOG performance, baseline PSA and LDH, Gleason score at diagnosis, and visceral disease at screening). A pre-specified follow-up rPFS analysis based on the investigator assessment of radiographic progression demonstrated a statistically significant improvement between the treatment groups with a 69.3% reduction in risk of radiographic progression or death [HR = 0.31 (95% CI: 0.27, 0.35), p < 0.0001]. The median rPFS was 19.7 months in the enzalutamide group and 5.4 months in the placebo group.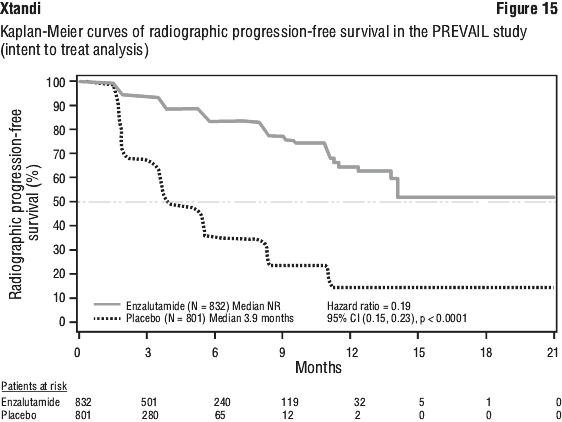 At the time of the primary analysis there were 1633 patients randomised.
At the time of the primary analysis there were 1633 patients randomised.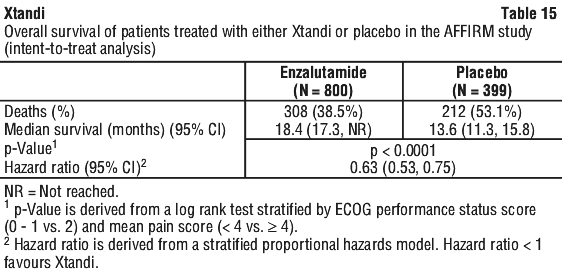
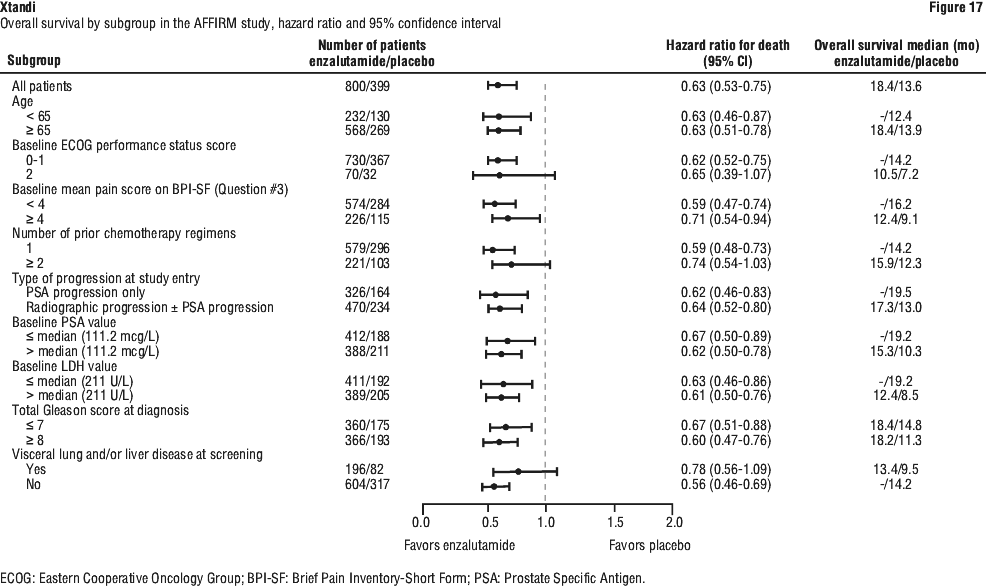 In addition to the observed improvement in overall survival, key secondary endpoints (radiographic progression-free survival, and time to first skeletal-related event) favoured Xtandi and were statistically significant after adjusting for multiple testing.
In addition to the observed improvement in overall survival, key secondary endpoints (radiographic progression-free survival, and time to first skeletal-related event) favoured Xtandi and were statistically significant after adjusting for multiple testing. Chemical name: 4-{3-[4-cyano-3-(trifluoromethyl)phenyl]- 5,5-dimethyl-4-oxo-2-thioxoimidazolidin-1-yl}-2-fluoro- N-methylbenzamide.
Chemical name: 4-{3-[4-cyano-3-(trifluoromethyl)phenyl]- 5,5-dimethyl-4-oxo-2-thioxoimidazolidin-1-yl}-2-fluoro- N-methylbenzamide.