What is in this leaflet
This leaflet answers some common questions about Zelboraf tablets.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Zelboraf against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Zelboraf is used for
Zelboraf contains the active ingredient vemurafenib.
Zelboraf belongs to a group of medicines called anti-neoplastic (or anti-cancer) agents.
Zelboraf is used to treat metastatic melanoma (a type of skin cancer that has spread to other parts of the body). It can only be used if your melanoma has a change (mutation) in the BRAF gene. Your doctor will have tested you for this gene. This change has been shown to be involved in the development of melanoma.
Zelboraf works by targeting proteins made from the BRAF gene to slow down the development of your cancer.
Ask your doctor if you have any questions about why Zelboraf has been prescribed for you.
Zelboraf is not addictive.
This medicine is available only with a doctor's prescription.
Before you take Zelboraf
When you must not take it
Do not take Zelboraf if:
- you are allergic (hypersensitive) to vemurafenib or any ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- the package is torn or shows signs of tampering
If the package is damaged, return it to your pharmacist for disposal.
- the expiry date (EXP) printed on the pack has passed.
If you take this medicine after the expiry date has passed, it may not work as well.
If you are not sure if you should start taking Zelboraf, talk to your doctor.
Before you start to take it
Tell your doctor if:
- you have a heart disorder,
such as abnormal electrical signal called "prolongation of the QT interval". Your doctor will test to check that your heart is working properly before and during your treatment with Zelboraf. If necessary, your doctor may decide to interrupt your treatment temporarily or stop it altogether.
- you have liver problems
Before and regularly during your treatment, your doctor will do some blood tests to monitor your liver function. If necessary, your doctor may decide to reduce your dose, interrupt your treatment temporarily or stop it altogether.
- you have kidney problems
Kidney problems may affect the activity of Zelboraf. Your doctor will do some blood tests to monitor your kidney function.
- you are pregnant or plan to become pregnant
Tell your doctor if you are pregnant or plan to become pregnant.
Zelboraf is not recommended for use during pregnancy. There is no information about the safety of Zelboraf in pregnant women. However if there is a need to take Zelboraf when you are pregnant your doctor will discuss the risks and benefits to you and the unborn baby.
Women of childbearing potential and men should use adequate contraception during treatment and for 6 months after the end of treatment with Zelboraf.
- you have been told that you have low blood levels of potassium, calcium, or magnesium in your blood
These levels may need to be corrected before treatment with Zelboraf begins.
- you have been previously diagnosed with other types of cancer
- you are breast-feeding or plan to breast-feed
It is not known whether Zelboraf passes into breast milk. Breast-feeding is not recommended during treatment with Zelboraf.
- you have previously received radiation treatment
- you are allergic to any other medicines, foods, dyes or preservatives
If you have not told your doctor about any of the above, tell him or her before you start taking Zelboraf.
Use in Children
Zelboraf is not recommended for use in children and adolescents. The safety and effectiveness in people younger than 18 years old have not been established.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought from a pharmacy, supermarket or health food shop. Tell your doctor if you are receiving treatment for any other medical conditions.
Zelboraf may interfere with some medicines. These include:
- ipilimumab, a medicine used to treat metastatic melanoma
- warfarin, a medicine used to prevent blood clots
- medicines that are mainly eliminated by or affect a metabolising protein called CYP3A4 such as;
- ketoconazole, itraconazole or voriconazole, medicines used to treat fungal infections
- atazanavir, saquinavir, ritonavir, nelfinavir or indinavir, medicines used to treat HIV infection
- phenytoin, carbamazepine or phenobarbital, medicines used to treat convulsions
- rifampicin, rifabutin, rifapentine, clarithromycin or telithromycin, medicines used to treat bacterial infections
- nefazodone or herbal medicines derived from St. John's Wort, used to treat depression
- some oral, injectable, or implantable contraceptives, for example "the Pill", used for birth control - medicines that are mainly eliminated by a metabolizing protein called CYP1A2 such as;
- caffeine, a stimulant
- olanzapine, clozapine, medicines used in treat schizophrenia and depression
- theophylline, a medicine used to treat asthma
- tizanidine, a medicine used as a muscle relaxant - medicines that influence, or are affected by, a protein called P-glycoprotein such as;
- verapamil, a medicine used to treat high blood pressure and angina (chest pain)
- cyclosporin, a medicine used to suppress the immune system
- dronedarone or digoxin, medicines used treat heart problems - medicines that may affect your heartbeat such as:
- those used to treat heart rhythm problems, such as amiodarone
- those used to treat depression, such as amitriptyline or imipramine
- those used to treat bacterial infections, such as azithromycin and clarithromycin
- ondansetron or domperidone, medicines used to treat nausea and vomiting. - medicines that influence, or are affected by, a protein called BCRP such as;
- methotrexate, medicine used to treat arthritis and some types of cancer
- mitoxantrone, a medicine used to treat some types of cancer
- rosuvastatin, a medicine used to lower high cholesterol
These medicines may be affected by Zelboraf, or may affect how well Zelboraf works. You may need to use different amounts of your medicine, or you may need to take different medicines. Your doctor will advise you.
This is not a complete list. Zelboraf may interfere with other medicines. Please tell your doctor or pharmacist if you are taking other medicines.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking Zelboraf.
Ask your doctor or pharmacist if you are not sure about this list of medicines.
How to take Zelboraf
Always take Zelboraf exactly as your doctor has told you. You should check with your doctor if you are not sure.
How much to take
The normal dose of Zelboraf is 4 tablets twice a day. Take 4 tablets in the morning, then 4 tablets in the evening, about 12 hours later.
If you experience side effects, your doctor may need to lower the dose to carry on your treatment.
Always take Zelboraf exactly as your doctor has told you. Your doctor's directions may differ from the information in this leaflet.
How to take it
Swallow the tablets whole with a glass of water.
Take Zelboraf on an empty stomach either at least 1 hour before or at least 2 hours after a meal in the morning and evening. Food may interfere with the absorption of Zelboraf.
Do not chew or crush the tablets.
How long to take it
How long you will be treated with Zelboraf depends on how you are responding to treatment. Your doctor will discuss this with you.
Continue taking Zelboraf until your doctor tells you to stop.
If you forget to take it
If you forget a dose take your dose as soon as you remember it.
If it is less than 4 hours before your next dose, skip the missed dose. Then take the next dose at the usual time.
Do not take a double dose to make up for a forgotten dose. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor, nurse or pharmacist.
If you have trouble remembering when to use your medicine, ask your pharmacist for some hints.
If you vomit after taking it
If you vomit after taking Zelboraf, do not take the same dose again. Then take the next dose at the usual time.
If you use too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (Australia - telephone 13 11 26; New Zealand - telephone 0800 764 766) for advice or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have used too much Zelboraf, even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Taking too much Zelboraf may make some side effects more likely or more severe.
Keep telephone numbers for these places handy.
While you are taking Zelboraf
Things you must do
Use Zelboraf exactly as your doctor has prescribed.
If you are about to be started on any new medicine or start radiation treatment, remind your doctor and pharmacist that you are taking Zelboraf.
Tell all doctors, dentists and pharmacists who are treating you that you are receiving Zelboraf.
Tell your doctor if you notice changes in your skin such as;
- a new wart
- a skin sore or reddish bump
- a sore that bleeds or does not heal.
Regularly during treatment and for up to 6 months after your treatment, your doctor needs to check your skin for a type of cancer called cutaneous squamous cell carcinoma. There is a high risk of developing these skin cancers while taking Zelboraf.
In clinical trials, one in four patients developed these skin cancers. These skin cancers may develop as early as the first month of Zelboraf treatment. Factors that increase your risk of developing these cancers are being 65 years old or older, prior skin cancer and long-term sun exposure.
Usually, this change appears on sun damaged skin, remains local and can be cured by surgical removal. If your doctor finds this type of skin cancer, they may treat it or may send you to another doctor for treatment.
In addition, check your skin and tell your doctor right away about any changes. You may develop new melanomas while taking Zelboraf. These lesions are usually removed by surgery and you can continue your treatment.
Avoid going out in the sun. If you are taking Zelboraf you may become more sensitive to sunlight and get symptoms of sunburn (such as redness, itching, swelling and blistering) more easily or get sunburns that can be severe.
To help protect against sunburn, if you do plan to go into the sun:
- wear clothing which protects your skin, including head, face, arms and legs
- use a broad spectrum (UVA/UVB) sunscreen and lip balm (minimum of SPF 30+, re-applied every 2 - 3 hours)
Tell your doctor if you become pregnant while using Zelboraf.
Women of childbearing potential and men should use adequate contraception during treatment and for 6 months after the end of treatment with Zelboraf.
Tell your doctor if you are breast-feeding while being treated with Zelboraf.
Tell your doctor if, for any reason, you have not used Zelboraf exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Tell your doctor if you feel Zelboraf is not helping your condition.
Be sure to keep all of your appointments with your doctor so that your progress can be checked. Your doctor may perform tests to help guide your treatment.
Things you must not do
Do not stop taking Zelboraf or change the dose without first checking with your doctor.
Do not let yourself run out of medicine over the weekend or on holidays.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting a pharmacist.
Do not give Zelboraf to anyone else even if they have the same condition as you.
You should not breast-feed your infant during treatment with Zelboraf. It is not known whether Zelboraf crosses into human breast milk.
Things to be careful of
Be careful driving or operating machinery until you know how Zelboraf affects you.
Zelboraf may affect the ability to drive or operate machinery due to side effects, which may be experienced while taking Zelboraf, as such
- feeling tired (fatigue)
- dizziness
- eye problems.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are receiving Zelboraf.
Zelboraf helps many people with metastatic melanoma but it may have unwanted side effects.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
If you are over 65 years of age you may have an increased chance of getting side effects.
Do not be alarmed by the following list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- red skin rashes, itching, dry or scaly skin, and hardened or thickened areas of the skin
- painful red lumps or patches of skin that may appear darker or harder than usual
- skin problems including warts
- sunburn
- increased sensitivity to light
- loss of appetite and weight loss
- headache
- changes in the way things taste
- drooping eyelid and sagging muscles on one side of the face caused by a paralysed nerve in the face (often reversible)
- tingling, burning feelings, or pain in your hands or feet
- diarrhoea
- constipation
- hair loss
- joint, muscle, or back pain
- unusual weakness or lack of energy
- feeling sick (nausea), vomiting
- feeling tired (fatigue)
- fever
- excess fluid (swelling), usually in the legs
- cough
- frequent infections such as fever, severe chills, sore throat or mouth ulcers
- thickening or appearance of visible cords, bands or lumps in the palm of one or both hands or feet
These are the more common side effects of Zelboraf.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- difficulty breathing, chest tightness or wheezing
- swelling of the face, lips or mouth
- severe skin rash, itching, hives
- severe blisters or bleeding of your lips, mouth, nose, or eyes
- a severe skin reaction starting with painful red areas, then large blisters and ends with peeling of layers of skin. This is accompanied by fever and chills, aching muscles and generally feeling unwell.
- fever associated with a mild to severe skin rash
- severe light-headedness or dizziness or feel your heart is beating irregularly or fast
- problems with your eyes or eyesight such as blurred or altered vision, irritated eyes, eye pain or redness
- yellowing of the skin and eyes
- light coloured bowel motions
- dark coloured urine
- severe upper stomach pain, often with nausea and vomiting (signs of inflammation of the pancreas)
These are serious side effects. You may need urgent medical attention.
Tell your doctor as soon as possible if you notice any changes in your skin.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Ask your doctor or pharmacist if you don't understand anything in this list.
After using Zelboraf
Storage
Keep Zelboraf in a cool dry place where the temperature stays below 30 °C.
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the blister pack they may not keep well.
Do not store Zelboraf, or any other medicine, in a bathroom or near a sink.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
Keep Zelboraf where young children cannot reach it. A locked cupboard at least 1.5 metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking Zelboraf, or the tablets have passed their expiry date, ask your pharmacist what to do with any tablets that are left over.
Product Description
Availability
Zelboraf 240 mg film-coated tablets are available in packs of 56 tablets (7 blisters of 8 tablets).
What it looks like
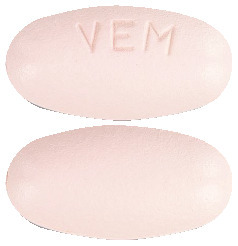
Zelboraf tablets are oval, biconvex, pinkish white to orange white tablets with "VEM" engraved on one side.
Ingredients
Active ingredient -
vemurafenib
Inactive ingredients
- croscarmellose sodium
- colloidal anhydrous silica
- magnesium stearate
- hydroxypropylcellulose
- hypromellose acetate succinate
The film-coating contains:
- polyvinyl alcohol
- titanium dioxide CI77891
- macrogol 3350
- talc, purified
- iron oxide red CI77491
Distributor
Zelboraf is distributed in Australia by:
Roche Products Pty Limited
ABN 70 000 132 865
Level 8, 30-34 Hickson Road
Sydney NSW 2000
AUSTRALIA
Medical enquiries: 1800 233 950
Zelboraf is distributed in New Zealand by:
Roche Products (New Zealand) Limited
PO Box 109113 Newmarket
Auckland 1149
NEW ZEALAND
Medical enquiries: 0800 276 243
Please check with your pharmacist for the latest Consumer Medicine Information.
Australian Registration Number
AUST R 183674
This leaflet was prepared on 25 March 2020
Published by MIMS May 2020



 Creatinine changes from baseline in the Phase III clinical study are summarised in Table 5.
Creatinine changes from baseline in the Phase III clinical study are summarised in Table 5.

 This inhibitory effect was confirmed in the ERK phosphorylation and cellular anti-proliferation assays in available melanoma cell lines expressing V600-mutant BRAF. In cellular anti-proliferation assays the inhibitory concentration 50 (IC50) against V600 mutated cell lines (V600E, V600R, V600D and V600K mutated cell lines) ranged from approximately 0.015 to 1 microM whereas the IC50 against BRAF wild type cell lines were > 10 microM.
This inhibitory effect was confirmed in the ERK phosphorylation and cellular anti-proliferation assays in available melanoma cell lines expressing V600-mutant BRAF. In cellular anti-proliferation assays the inhibitory concentration 50 (IC50) against V600 mutated cell lines (V600E, V600R, V600D and V600K mutated cell lines) ranged from approximately 0.015 to 1 microM whereas the IC50 against BRAF wild type cell lines were > 10 microM.
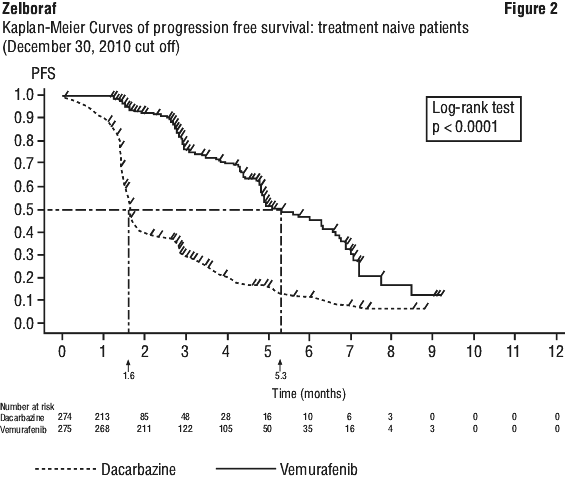
 Table 9 and Figure 2 show the progression-free survival in treatment-naïve patients with BRAF V600 mutation positive melanoma.
Table 9 and Figure 2 show the progression-free survival in treatment-naïve patients with BRAF V600 mutation positive melanoma.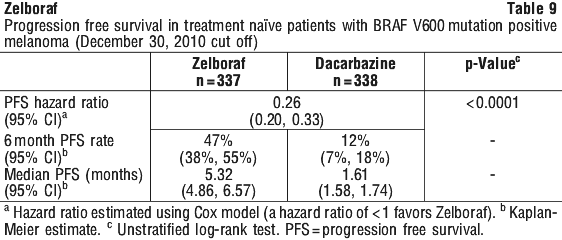


 Vemurafenib is designated chemically as N-{3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b] pyridin-3-carbonyl]-2,4-difluorophenyl} propane-1-sulfonamide.
Vemurafenib is designated chemically as N-{3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b] pyridin-3-carbonyl]-2,4-difluorophenyl} propane-1-sulfonamide.