1 Name of Medicine
Zilucoplan tetrasodium.
2 Qualitative and Quantitative Composition
Zilucoplan is a 15 amino acid, synthetic macrocyclic peptide.
Three pre-filled syringe presentations are available:
a pre-filled syringe of 0.416 mL contains zilucoplan tetrasodium, equivalent to 16.6 mg of zilucoplan;
a pre-filled syringe of 0.574 mL contains zilucoplan tetrasodium, equivalent to 23.0 mg of zilucoplan;
a pre-filled syringe of 0.810 mL contains zilucoplan tetrasodium, equivalent to 32.4 mg of zilucoplan.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
The solution is sterile, clear to slightly opalescent and colourless, free of visible particles.
4.1 Therapeutic Indications
Zilbrysq is indicated as an add-on to standard therapy for the treatment of generalised myasthenia gravis (gMG) in adult patients who are anti-acetylcholine receptor (AChR) antibody positive.
4.2 Dose and Method of Administration
Before starting therapy with Zilbrysq, patients must be vaccinated against Neisseria meningitidis. If treatment with Zilbrysq needs to start less than 2 weeks after vaccination against meningococcal infection, the patient must receive appropriate prophylactic antibiotic treatment until 2 weeks after the first vaccination dose (see Section 4.3 Contraindications; Section 4.4 Special Warnings and Precautions for Use).
The recommended dose corresponds to approximately 0.3 mg/kg, given as a once daily subcutaneous injection.
Zilbrysq is for single use in one patient only. Discard any residue.
Table 1 indicates the total daily dose of Zilbrysq per body weight range (see Section 5.2 Pharmacokinetic Properties).
 There is limited experience with patients below 43 kg and above 150 kg. These patients should receive the lowest and the highest dose, respectively.
There is limited experience with patients below 43 kg and above 150 kg. These patients should receive the lowest and the highest dose, respectively.
The daily dose should be administered approximately at the same time every day.
Missed dose.
If the Zilbrysq dose is missed, administer the dose as soon as possible on the same day and then resume dosing at the scheduled time on the following day. Do not administer more than one dose per day.
Special populations.
No dose adjustment is required based on age, gender, race or ethnicity, or in patients with renal impairment and/or mild to moderate hepatic impairment (see Section 4.4 Special Warnings and Precautions for Use; Section 5.2 Pharmacokinetic Properties).
Method of administration.
Zilbrysq is administered by subcutaneous injection.
Suitable injection sites include front of the thighs, the abdomen and the back of the upper arms.
Injection sites should be rotated and injections should not be given in areas where the skin is tender, erythematous, bruised, indurated or where the skin has scars or stretch marks.
Administration should be performed by an individual who has been trained in the correct injection techniques for Zilbrysq and following the detailed instructions for use within the pack.4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients (see Section 6.1 List of Excipients).
Patients who are not currently vaccinated against Neisseria meningitidis, unless they receive appropriate prophylactic antibiotic treatment until 2 weeks after the first vaccination dose (see Section 4.4 Special Warnings and Precautions for Use).
Patients with unresolved Neisseria meningitidis infection.
4.4 Special Warnings and Precautions for Use
Neisseria infections.
Meningococcal infection.
Due to its mechanism of action, the use of Zilbrysq may increase the patient's susceptibility to infections with Neisseria meningitidis. As a precautionary measure, all patients must be vaccinated against meningococcal infections, at least 2 weeks prior to the start of treatment with Zilbrysq.
If urgent treatment with Zilbrysq needs to start less than 2 weeks after vaccination against meningococcal infections, the patient must receive appropriate prophylactic antibiotic treatment until 2 weeks after the first vaccination dose. As meningococcal vaccines reduce but do not completely eliminate the risk of meningococcal infections, consideration should be given to official guidance on the appropriate use of antibiotics. Prescribers and patients should discuss the potential role of ongoing prophylactic antibiotic use.
Vaccines against serogroups A, C, Y, W, and where available, serogroup B, are recommended for preventing the commonly pathogenic meningococcal serogroups. Vaccination, revaccination (boosters) and prophylactic antibiotic treatment should occur according to current relevant guidelines.
During treatment with Zilbrysq, patients should be monitored for signs and symptoms of meningococcal infection and evaluated immediately if infection is suspected. In case of suspected meningococcal infection, appropriate measures such as treatment with antibiotics and discontinuation of treatment with Zilbrysq, should be taken until meningococcal infection can be ruled out. Patients should be instructed to seek immediate medical advice if signs or symptoms of meningococcal infections occur.
No cases of meningococcal infections were reported in the Phase 2 and Phase 3 placebo-controlled studies with Zilbrysq in gMG.
Prescribers should be familiar with the educational materials for the management of meningococcal infections and provide a patient alert card and patient/carer guide to patients treated with zilucoplan.
Other Neisseria infections.
In addition to Neisseria meningitidis, patients treated with Zilbrysq may also be susceptible to infections with other Neisseria species, such as gonococcal infections. Patients should be informed on the importance of gonorrhoea prevention and treatment.
Other infections.
Zilbrysq blocks terminal complement activation; therefore, patients may have increased susceptibility to infections, especially with encapsulated bacteria. During treatment with Zilbrysq, patients should be monitored for signs and symptoms of infections and evaluated if infection is suspected.
Pancreatic enzymes increased.
Elevations of lipase and/or amylase were observed, of which some were marked (CTCAE grade 3 and 4). These were transient and rarely led to treatment discontinuation. Although a causal relationship with pancreatitis or other pancreas pathologies (cysts, masses) has not been identified, cases of both were reported amongst patients treated with zilucoplan. In the evaluation of abdominal pain with suspicion of pancreatitis, appropriate investigations, including measurement of amylase and lipase should be undertaken.
Use in hepatic impairment.
No dose adjustment is required for patients with mild and moderate hepatic impairment. There are no data on patients with severe hepatic impairment (see Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
No dose adjustment is required for patients with renal impairment. There are no data on patients requiring dialysis (see Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
No dose adjustment is required in elderly patients.
Paediatric use.
The safety and efficacy of Zilbrysq in children below the age of 18 years have not been established. No data are available.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No clinical interaction studies have been performed. Based on results from in vitro testing, clinically relevant interactions are not expected between zilucoplan and an inhibitor or inducer of major CYP enzymes or transporters.
Zilucoplan is not a substrate of major CYP enzymes or transporters (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 3A4, or P-gp, BCRP, OATP1B1 or OATP1B3).
The potential of zilucoplan to inhibit CYP enzymes (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A and 4F2) and UGTs (1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15) or transporters (P-gp, BCRP, BSEP, MRP2, MRP3, MATE1, MATE2-k, NTCP, OCT1, OCT2, OAT1, OAT3, OATP1B1, and OATP1B3) was evaluated in vitro. In addition, the potential of CYP induction of CYP1A2, 2B6 and CYP3A4 by zilucoplan was evaluated. Based on the results, zilucoplan will not inhibit or induce these major drug metabolising enzymes (CYPs and UGTs) and transporters in a clinically relevant manner. Some inhibition of MRP3 was observed, the relevance of which is unknown.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
The effect of Zilbrysq on human fertility has not been evaluated.
In a monkey male fertility study, minimal to slight testicular germ cell degeneration/depletion was observed at all subcutaneous doses (greater than or equal to 1 mg/kg/day, resulting in exposures based on AUC greater than or equal to 2 times the clinical AUC at the maximum human recommended dose) at the end of the 13-week dosing period and after the 8-week recovery period, but severity did not increase with dose. The findings in non-human primates are of uncertain clinical relevance as the underlying mechanism is unknown.
Zilucoplan-induced effects on female fertility was not investigated in animals.
(Category D)
There are no data from the use of Zilbrysq in pregnant women. Zilbrysq is not recommended during pregnancy and in women of childbearing potential not using contraception.
Subcutaneous administration of zilucoplan (0, 1, 2, or 4 mg/kg/day) to pregnant monkeys throughout gestation resulted in an increase in embryofetal death at all doses, in the absence of maternal toxicity. A no effect dose for adverse developmental effects in monkeys was not identified. The lowest dose tested was associated with maternal exposures (AUC) similar to that in humans at the maximum recommended human dose.
Data collected from an ex vivo human placental transfer model suggests a low transfer rate of zilucoplan (0.5-1.0%) in the fetal compartment. The clinical relevance of these data in human pregnancies is unknown.
There are no data on the presence of Zilbrysq in human milk, the effects on the breastfed infant, or the effects on milk production.
A decision must be made whether to discontinue breastfeeding or to discontinue Zilbrysq therapy taking into account the benefit of breastfeeding for the child, as well as any potential adverse effects, and the benefit of therapy for the woman based on their underlying condition.4.7 Effects on Ability to Drive and Use Machines
Zilbrysq has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
A total of 115 patients have been treated with zilucoplan in placebo-controlled clinical studies in gMG, representing 26.4 patient-years of exposure.
Table 2 presents the treatment emergent adverse events reported in at least 5% of Zilbrysq-treated patients and more frequently than the placebo in the 12-week controlled period of the Phase 3 clinical trial in gMG.
 Very common adverse reactions were injection site reactions and upper respiratory tract infections reported in ≥ 10% of patients. Common adverse reactions were diarrhoea, amylase increased, lipase increased, and morphoea (localised scleroderma) reported in > 5% of patients. Uncommon adverse reactions were blood eosinophils increased reported in < 1% of patients.
Very common adverse reactions were injection site reactions and upper respiratory tract infections reported in ≥ 10% of patients. Common adverse reactions were diarrhoea, amylase increased, lipase increased, and morphoea (localised scleroderma) reported in > 5% of patients. Uncommon adverse reactions were blood eosinophils increased reported in < 1% of patients.
Description of selected adverse reactions.
Injection site reactions.
Most common terms were injection site bruising, pain, nodule, pruritus and haematoma. All cases were non-serious, mild or moderate in severity, and less than 3% of events led to treatment discontinuation. In pooled placebo-controlled studies, injection site reactions were reported in 25.2% of patients treated with zilucoplan and in 15.5% of patients treated with placebo.
Upper respiratory tract infections.
Most common terms were nasopharyngitis, upper respiratory tract infection and sinusitis. More than 95% of the cases were non-serious, mild or moderate in severity and did not lead to treatment discontinuation. In pooled placebo-controlled studies, upper respiratory tract infections were reported in 13.0% of patients treated with zilucoplan and in 7.8% of patients treated with placebo.
Blood eosinophils increased.
Elevations of blood eosinophils were observed in some subjects, of which some were marked. The majority peaked approximately 2 months after starting zilucoplan. These were transient, not leading to treatment discontinuation and not associated with clinically relevant organ dysfunction.
Morphoea.
Cases of morphoea (common, 1-10%) were observed after long-term treatment during the open-label extension study; the longest exposure to Zilbrysq in this study (RAISE-XT) was more than 4 years. The majority of the cases had a time to onset longer than one year after start of treatment, were mild or moderate in severity and did not lead to treatment discontinuation. The incidence of morphoea observed with zilucoplan appears to be higher than the expected background incidence in the MG population.
Immunogenicity.
As with all therapeutic peptides, there is a potential for immunogenicity with zilucoplan. The detection of anti-drug antibody (ADA) formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of ADA positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to zilucoplan with the incidence of antibodies to other products may be misleading.
In the Phase 3 clinical trial (MG0010), the patients were tested for ADA positivity and anti-PEG (polyethylene glycol) antibody positivity. A total of 2 patients (2.3%) each in the zilucoplan 0.3 mg/kg and placebo group were ADA positive. A total of 8 patients (9.3%) in the zilucoplan 0.3 mg/kg group and 6 patients (6.8%) in the placebo group were anti-PEG positive, respectively. Antibody titres were low and there was no evidence of an association between positive ADA status or positive anti-PEG status and the incidence of adverse events.
Patients with other diseases.
Supportive safety data were obtained from 7 completed clinical studies that enrolled 225 patients exposed to zilucoplan in disease populations other than gMG (Immune-mediated necrotizing myopathy, COVID-19 associated respiratory syndrome and paroxysmal nocturnal hemoglobinuria). No additional ADR were identified in patients with these diseases compared to patients with gMG.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.4.9 Overdose
Limited experience with doses higher than the recommended dose of zilucoplan is available from clinical trials in humans.
In a healthy volunteer study where 32 participants were exposed to supratherapeutic doses of 0.6 mg/kg, administered subcutaneously for up to 7 days, safety data were consistent with the safety profile of the recommended dose.
In cases of overdosage, it is recommended that patients are monitored closely for any adverse effects, and appropriate supportive measures should be instituted immediately.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
In gMG patients, binding of anti-AChR auto-antibodies (mainly IgG1 and IgG3) to AChR results in uncontrolled and inappropriate activation of the classical complement pathway. The immune complex formed by the autoantibody-antigen complex activates the C1 component of the classical complement pathway. This leads to a series of enzymatic cleavage steps, culminating in the cleavage of C5 into C5a and C5b and deposition of the cytolytic membrane attack complex (C5b-9, MAC) on the post-synaptic membrane of the neuromuscular junction and subsequent injury to the neuromuscular endplate, leading to failure of neuromuscular transmission.
Zilucoplan inhibits the effects of C5 through a dual mechanism of action. It specifically binds to complement protein C5, thereby inhibiting its cleavage by the C5 convertase to C5a and C5b, which results in a downregulation of the assembly and cytolytic activity of the MAC. Additionally, by binding to the C5b moiety of C5, zilucoplan sterically hinders binding of C5b to C6, which prevents the subsequent assembly and activity of the MAC, should any C5b be formed.
Pharmacodynamic effects.
The pharmacodynamic effect of zilucoplan was analysed through the ability of inhibiting ex vivo, complement-induced sheep red blood cell (sRBC) lysis.
In the Phase 2 study in gMG (MG0009), patients received zilucoplan 0.3 mg/kg, zilucoplan 0.1 mg/kg or placebo daily for 12 weeks. There was rapid complement inhibition within 1-3 hours and at Week 12, complement inhibition was 95.7% in patients receiving zilucoplan 0.3 mg/kg, compared with 81.8% in patients receiving zilucoplan 0.1 mg/kg.
In the Phase 3 study in gMG (MG0010), patients received zilucoplan 0.3 mg/kg or placebo daily for 12 weeks. Similarly, complete complement inhibition of 97.5% could be seen from Week 1 through Week 12 with zilucoplan.
This effect was maintained in the Phase 3 open-label extension study MG0011 where complement inhibition at Week 12 was 97.3% in patients who were treated with zilucoplan in MG0009 or MG0010 and 95.9% in patients who had been treated with placebo in MG0009 or MG0010 and switched to zilucoplan in the open-label extension.
Data from the Phase 2 and Phase 3 studies demonstrate rapid, complete and sustained complement inhibition with zilucoplan 0.3 mg/kg.
Clinical trials.
The safety and efficacy of zilucoplan were evaluated in a 12-week multicentre, randomised, double-blind placebo-controlled study MG0010 (RAISE) and the open-label extension study MG0011 (RAISE-XT).
Study MG0010 (RAISE).
A total of 174 patients were enrolled, who were at least 18 years of age, had anti-AChR antibody positive gMG, MGFA Class II-IV (mild to severe), a Myasthenia Gravis-Activities of Daily Living (MG-ADL) Score of ≥ 6 and a Quantitative Myasthenia Gravis (QMG Score) of ≥ 12 (see Table 3).
Patients were treated with zilucoplan 0.3 mg/kg once daily or placebo. The 0.3 mg/kg dose was given according to the same weight categories as the proposed clinical use. Stable standard of care (SOC) therapy was allowed.
The primary endpoint was the change from baseline (CFB) to Week 12 in MG-ADL total score. MG-ADL is an 8-item patient reported outcome measure assessing impact of gMG on daily function of signs and symptoms. The total score is the sum of the 8 individual scores and ranges from 0 to 24, with higher scores indicating more severe impact of gMG on daily function.
Key secondary endpoints were the CFB to Week 12 in QMG total score, in Myasthenia Gravis Composite (MGC) total score and in MG Quality of Life (MG-QoL15r) total score.
QMG is a 13-item clinician reported outcome measure assessing muscle weakness. The total score is the sum of the 13 individual scores and ranges from 0 to 39, with higher scores indicating more severe weakness.
MGC is a 10-item gMG composite (reported by clinician and patient) outcome measure to evaluate gMG clinical status. The range of total scores is 0 to 50, with higher scores indicating more severe impairment due to gMG.
MG-QoL15r is a 15-item patient reported outcome measure assessing the impact of gMG on patient's quality of life. The total score ranges from 0 to 30, with higher scores indicating greater impairment of QoL.
All 4 outcome measures are validated in gMG.
The majority of study participants received treatment for gMG at baseline which included parasympathomimetics (84.5%), systemic corticosteroids (63.2%) and immunosuppressants (51.1%). The immunosuppressants used were mainly azathioprine and mycophenolate. 13.2% of patients were on intravenous immunoglobulin (IVIG) at baseline.
The mean weight of participants was 89.1 kg and mean BMI was 31 kg/m2 and these were balanced between the groups.
 The treatment effect in the zilucoplan group for all 4 endpoints started rapidly at Week 1, further increased to Week 4 and was sustained through Week 12.
The treatment effect in the zilucoplan group for all 4 endpoints started rapidly at Week 1, further increased to Week 4 and was sustained through Week 12.
At Week 12, a clinically meaningful and highly statistically significant improvement in the primary endpoint, MG-ADL total score (Figure 1) was observed for zilucoplan versus placebo.
 For the key secondary endpoints, at Week 12, a clinically meaningful and highly statistically significant improvement in QMG total score (Figure 2) was observed for zilucoplan versus placebo. At Week 12, a clinically meaningful and statistically significant improvement in MGC total score (Figure 3) and a statistically significant improvement in MG-QoL15r total score (Figure 4) was observed for zilucoplan versus placebo.
For the key secondary endpoints, at Week 12, a clinically meaningful and highly statistically significant improvement in QMG total score (Figure 2) was observed for zilucoplan versus placebo. At Week 12, a clinically meaningful and statistically significant improvement in MGC total score (Figure 3) and a statistically significant improvement in MG-QoL15r total score (Figure 4) was observed for zilucoplan versus placebo.


 For the secondary endpoints, all p-values were based on the pre-defined hierarchy. At Week 12, 73.1% of the patients in the zilucoplan group were MG-ADL clinical responders (defined as having at least a 3-point decrease in MG-ADL score) without rescue therapy, vs. 46.1% in the placebo group (statistically significant, p < 0.001). 58.0% of the patients in the zilucoplan group were QMG clinical responders (defined as having at least a 5-point decrease in QMG score) without rescue therapy, vs. 33.0% in the placebo group (statistically significant, p=0.0012). The proportion of clinical responders at higher response thresholds was consistently greater for zilucoplan compared with placebo. 14.0% of the patients in the zilucoplan group achieved Minimal Symptom Expression (MG-ADL score of 0 or 1; MSE) without rescue therapy, vs. 5.8% in the placebo group (p=0.0885).
For the secondary endpoints, all p-values were based on the pre-defined hierarchy. At Week 12, 73.1% of the patients in the zilucoplan group were MG-ADL clinical responders (defined as having at least a 3-point decrease in MG-ADL score) without rescue therapy, vs. 46.1% in the placebo group (statistically significant, p < 0.001). 58.0% of the patients in the zilucoplan group were QMG clinical responders (defined as having at least a 5-point decrease in QMG score) without rescue therapy, vs. 33.0% in the placebo group (statistically significant, p=0.0012). The proportion of clinical responders at higher response thresholds was consistently greater for zilucoplan compared with placebo. 14.0% of the patients in the zilucoplan group achieved Minimal Symptom Expression (MG-ADL score of 0 or 1; MSE) without rescue therapy, vs. 5.8% in the placebo group (p=0.0885).
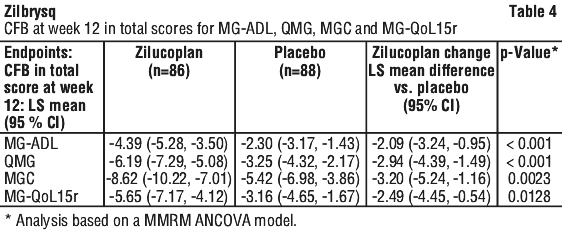
Table 4 presents the CFB at week 12 in the total scores for MG-ADL, QMG, MGC and MG-QoL15r.
 At Week 12, the cumulative portion of patients that needed rescue therapy was lower in the zilucoplan group (5%) compared with the placebo group (12%). Separation of zilucoplan from placebo was first observed by Week 2 (Day 15); this separation was sustained through Week 12 (Day 84). This only shows a trend and is non-significant.
At Week 12, the cumulative portion of patients that needed rescue therapy was lower in the zilucoplan group (5%) compared with the placebo group (12%). Separation of zilucoplan from placebo was first observed by Week 2 (Day 15); this separation was sustained through Week 12 (Day 84). This only shows a trend and is non-significant.
Treatment with zilucoplan resulted in consistently greater decreases from baseline in the respective subgroups (refractory status, age, gender, ethnicity, disease duration or severity, baseline MG-ADL and QMG scores and prior treatment with steroids, plasma exchange (PLEX), IVIG, subcutaneous immunoglobulin (SCIG) or immunosuppressants) compared with placebo for the primary and key secondary endpoints.
Study MG0011 (RAISE-XT).
Two hundred patients who completed the placebo-controlled Phase 2 (MG0009) or Phase 3 (MG0010) studies entered the open-label extension study MG0011 and were treated with zilucoplan 0.3 mg/kg daily. MG0011 was primarily a safety and tolerability study which was not randomized or placebo-controlled, therefore efficacy is inferred. Efficacy endpoints were the CFB in MG-ADL, QMG, MGC and MG-QoL15r score at Week 12 of the open-label extension period. The results of MG0011 demonstrated sustained efficacy through to Week 60.
Figure 5 shows the mean changes from double-blind study baseline to Week 48 of the open-label extension for total MG-ADL score. See Table 5.


5.2 Pharmacokinetic Properties
The pharmacokinetic (PK) properties of zilucoplan and the major circulating metabolites (RA102758 and RA103488) have been evaluated in healthy adult subjects and in patients with gMG.
A population PK analysis showed that zilucoplan PK was not time and dose dependent. At the therapeutic dose of 0.3 mg/kg, the PK of zilucoplan is in the linear dose range.
Absorption.
Following single and multiple daily subcutaneous administration of zilucoplan 0.3 mg/kg in healthy subjects, zilucoplan reached peak plasma concentrations generally between 3 to 6 hours post-dose.
In study MG0010 in patients with gMG, after daily repeated subcutaneous administration of zilucoplan 0.3 mg/kg, plasma concentrations of zilucoplan were consistent, with steady state trough concentrations being reached by Week 4 of zilucoplan treatment and maintained through Week 12. The ADA and anti-PEG antibody status of patients treated with zilucoplan did not affect zilucoplan concentrations.
Exposures after subcutaneous administration of single zilucoplan (0.3 mg/kg) doses in the abdomen, thigh, or upper arm were comparable.
Distribution.
Zilucoplan and its 2 major metabolites are highly bound to plasma proteins (> 99%). The mean volume of distribution for zilucoplan (Vc/F) using a population PK analysis is 3.51 L.
Metabolism.
In plasma, 2 major metabolites, RA103488 and RA102758 were detected. The formation of RA103488 is mainly due to cytochrome CYP450 4F2. RA103488 has pharmacological activity similar to zilucoplan but is present at a much lower concentration compared with zilucoplan. The contribution of RA103488 to pharmacological activity is low. RA102758 is pharmacologically inactive. Further, as a peptide, zilucoplan is expected to be degraded into small peptides and amino acids via catabolic pathways.
Excretion.
The mean plasma terminal elimination half-life was approximately 172 hours (7-8 days). The excretion of zilucoplan and its metabolites was measured in both urine and faeces and was negligible or < 1%.
Special populations.
Elderly.
Based on population PK analysis, age did not influence the PK of zilucoplan. No dose adjustment is required.
Renal impairment.
The effect of renal impairment on the PK of zilucoplan and its metabolites was studied in an open-label Phase 1 study, where a single dose of zilucoplan 0.3 mg/kg was administered to healthy subjects and subjects with severe renal impairment (creatinine clearance between 15 and < 30 mL/min).
Systemic exposure to zilucoplan and the major inactive metabolite RA102758 was not different in subjects with severe renal impairment compared to subjects with normal renal function. The exposure to the active metabolite RA103488 was approximately 1.5-fold higher in subjects with severe renal impairment compared to subjects with normal renal function.
Based on the PK results, no dose adjustment is required in patients with renal impairment.
Hepatic impairment.
The effects of moderate hepatic impairment (as defined by a Child-Pugh score between 7 and 9) on the PK of zilucoplan and its metabolites were studied in an open-label Phase 1 study, where a single dose of 0.3 mg/kg zilucoplan was administered to healthy subjects and subjects with moderate hepatic impairment.
Systemic exposure to zilucoplan, based on AUC0-last and AUC(0-inf), was 24% lower in subjects with moderate impaired liver function compared with healthy subjects, which was in line with a higher systemic and peak exposures of both metabolites in subjects with hepatic impairment compared with healthy subjects. Zilucoplan peak exposure as well as terminal half-life were comparable between both groups. Further pharmacodynamic analysis did not identify meaningful differences in complement levels or inhibition of complement activity between both groups. Based on these results, no dose adjustment is required in patients with mild and moderate hepatic impairment.
Racial and ethnic groups.
In a Phase 1 clinical study in healthy Caucasian and Japanese subjects, the PK profile of zilucoplan and its 2 major metabolites was compared following a single dose of 0.3 mg/kg and after multiple dosing of 0.3 mg/kg for 14 days. Results were generally similar between both groups. The population PK analysis demonstrated that there are no differences between the different race categories (Black/African American, Asian/Japanese, and Caucasians). No dose adjustment is required.
Gender.
In the population PK analysis, no difference in PK between genders was observed. No dose adjustment is required.
Weight.
Population PK analysis on data collected across studies in gMG showed that body weight significantly influences the PK of zilucoplan. Zilucoplan dosing is based on body weight categories (see Section 4.2 Dose and Method of Administration), no further dose adjustment is needed.
5.3 Preclinical Safety Data
Genotoxicity.
Zilucoplan was not genotoxic when tested in the in vitro mutagenicity test (Ames), the in vitro chromosomal aberration assay and in the in vivo micronucleus test in rat bone marrow cells.
Carcinogenicity.
No carcinogenicity studies were conducted with zilucoplan.6 Pharmaceutical Particulars
6.1 List of Excipients
The inactive ingredients are monobasic sodium phosphate monohydrate, dibasic sodium phosphate, sodium chloride and water for injections.
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.4 Special Precautions for Storage
Store in a refrigerator (2°C - 8°C).
Do not freeze.
Keep the pre-filled syringe in the original carton to protect from light.
Patients may store the Zilbrysq pre-filled syringe(s) at room temperature in the original carton (with protection from light) at up to 30°C for a single maximum period of 3 months. Once Zilbrysq has been stored at room temperature, it should not be placed back into the refrigerator and should be discarded if not used within the 3 month period or by the expiry date, whichever occurs first.
6.5 Nature and Contents of Container
Each pre-filled syringe is type I glass with a 29 G ½" thin wall needle closed with a grey fluoropolymer-laminated bromobutyl rubber plunger stopper. The needle is protected with a rigid needle shield consisting of a thermoplastic elastomer needle shield and a polypropylene rigid shield. The pre-filled syringe components are not made with natural rubber latex.
Each pre-filled syringe is pre-assembled with a needle safety device, a finger grip and a coloured plunger.
The pre-filled syringes are available as 3 different presentations, with different volumes to allow patients to receive the appropriate dose based on a weight range:
0.416 mL pre-filled syringe with rubine red plunger;
0.574 mL pre-filled syringe with orange plunger;
0.810 mL pre-filled syringe with dark blue plunger.
Pack size: 7 pre-filled syringes.
Multipack containing 28 (4 packs of 7) pre-filled syringes.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical name: zilucoplan tetrasodium.
Molecular formula and mass: the molecular formula of zilucoplan (free acid form) is C172H278N24O55 and its molecular weight is 3562.23 Da.
Chemical structure.
 The four sodium ions in the structure are shown associated with designated carboxylates, but they may be associated with any of the acidic groups in the molecule.
The four sodium ions in the structure are shown associated with designated carboxylates, but they may be associated with any of the acidic groups in the molecule.
CAS number.
1841136-73-9 (free acid).7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
